Este artículo necesita citas adicionales para su verificación . ( marzo de 2014 ) ( Aprenda cómo y cuándo eliminar este mensaje de plantilla ) |
.png){kind=link}
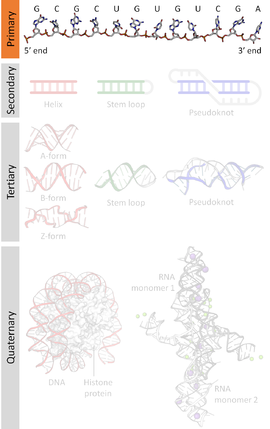
Una secuencia de ácido nucleico es una sucesión de bases representadas por una serie de un conjunto de cinco letras diferentes que indican el orden de los nucleótidos que forman los alelos dentro de una molécula de ADN (usando GACT) o ARN (GACU). Por convención, las secuencias se presentan normalmente desde el extremo 5 'hasta el extremo 3' . Para el ADN, se usa la hebra con sentido . Debido a que los ácidos nucleicos son normalmente polímeros lineales (no ramificados) , especificar la secuencia equivale a definir la estructura covalente de la molécula completa. Por esta razón, la secuencia de ácido nucleico también se denomina estructura primaria .
La secuencia tiene capacidad para representar información . El ácido desoxirribonucleico biológico representa la información que dirige las funciones de un ser vivo.
Los ácidos nucleicos también tienen una estructura secundaria y una estructura terciaria . La estructura primaria a veces se denomina erróneamente secuencia primaria . Por el contrario, no existe un concepto paralelo de secuencia secundaria o terciaria.
Nucleótidos [ editar ]
Los ácidos nucleicos consisten en una cadena de unidades unidas llamadas nucleótidos. Cada nucleótido consta de tres subunidades: un grupo fosfato y un azúcar ( ribosa en el caso del ARN , desoxirribosa en el ADN ) forman la columna vertebral de la cadena de ácido nucleico, y unido al azúcar es uno de un conjunto de bases nucleicas . Las nucleobases son importantes en el emparejamiento de bases de las cadenas para formar una estructura secundaria y terciaria de nivel superior , como la famosa doble hélice .
Las letras posibles son A , C , G y T , que representan las cuatro bases de nucleótidos de una cadena de ADN ( adenina , citosina , guanina , timina ) unidas covalentemente a una cadena principal de fosfodiéster . En el caso típico, las secuencias se imprimen contiguas entre sí sin espacios, como en la secuencia AAAGTCTGAC, leída de izquierda a derecha en la dirección de 5 'a 3' . Con respecto a la transcripción , una secuencia está en la cadena codificante si tiene el mismo orden que el ARN transcrito.
Una secuencia puede ser complementaria a otra secuencia, lo que significa que tienen la base en cada posición en la complementaria (es decir, A a T, C a G) y en el orden inverso. Por ejemplo, la secuencia complementaria de TTAC es GTAA. Si una hebra del ADN de doble hebra se considera la hebra con sentido, entonces la otra hebra, considerada la hebra antisentido, tendrá la secuencia complementaria a la hebra con sentido.
Notación [ editar ]
Comparación y determinación del% de diferencia entre dos secuencias de nucleótidos.
- ETIQUETA AA T CC GC
- ETIQUETA AA A CC CT
- Dadas las dos secuencias de 10 nucleótidos, alinéalas y compara las diferencias entre ellas. Calcule el porcentaje de similitud tomando el número de bases de ADN diferentes dividido por el número total de nucleótidos. En el caso anterior, hay tres diferencias en la secuencia de 10 nucleótidos. Por lo tanto, divida 7/10 para obtener el 70% de similitud y réstelo del 100% para obtener una diferencia del 30%.
Si bien A, T, C y G representan un nucleótido particular en una posición, también hay letras que representan ambigüedad que se usan cuando más de un tipo de nucleótido podría ocurrir en esa posición. Las reglas de la Unión Internacional de Química Pura y Aplicada ( IUPAC ) son las siguientes: [1]
| Símbolo [2] | Descripción | Bases representadas | Complemento | ||||
|---|---|---|---|---|---|---|---|
| A | Un denine | A | 1 | T | |||
| C | C ytosine | C | GRAMO | ||||
| GRAMO | G uanine | GRAMO | C | ||||
| T | T hymine | T | A | ||||
| U | U racil | U | A | ||||
| W | W EAK | A | T | 2 | W | ||
| S | S trong | C | GRAMO | S | |||
| METRO | un M ino | A | C | K | |||
| K | K eto | GRAMO | T | METRO | |||
| R | pu R ine | A | GRAMO | Y | |||
| Y | p Y rimidina | C | T | R | |||
| B | no A ( B viene después de A) | C | GRAMO | T | 3 | V | |
| D | no C ( D viene después de C) | A | GRAMO | T | H | ||
| H | no G ( H viene después de G) | A | C | T | D | ||
| V | no T ( V viene después de T y U) | A | C | GRAMO | B | ||
| norte | cualquier N ucleotide (no una brecha) | A | C | GRAMO | T | 4 | norte |
| Z | Z ero | 0 | Z | ||||
Estos símbolos también son válidos para el ARN, excepto cuando U (uracilo) reemplaza a T (timina). [1]
Además de la adenina (A), la citosina (C), la guanina (G), la timina (T) y el uracilo (U), el ADN y el ARN también contienen bases que se han modificado después de que se haya formado la cadena de ácido nucleico. En el ADN, la base modificada más común es la 5-metilcitidina (m5C). En el ARN, hay muchas bases modificadas, que incluyen pseudouridina (Ψ), dihidrouridina (D), inosina (I), ribotimidina (rT) y 7-metilguanosina (m7G). [3] [4] La hipoxantina y la xantina son dos de las muchas bases creadas a través de la presencia de mutágenos , ambas mediante desaminación (reemplazo del grupo amina por un grupo carbonilo). La hipoxantina se produce a partir de adenina y la xantina se produce a partir de guanina.. [5] De manera similar, la desaminación de la citosina produce uracilo .
Importancia biológica [ editar ]
En los sistemas biológicos, los ácidos nucleicos contienen información que es utilizada por una célula viva para construir proteínas específicas . La secuencia de nucleobases en una hebra de ácido nucleico es traducida por maquinaria celular en una secuencia de aminoácidos que forman una hebra de proteína. Cada grupo de tres bases, llamado codón , corresponde a un solo aminoácido, y existe un código genético específico por el cual cada posible combinación de tres bases corresponde a un aminoácido específico.
El dogma central de la biología molecular describe el mecanismo por el cual se construyen las proteínas utilizando la información contenida en los ácidos nucleicos. El ADN se transcribe en moléculas de ARNm , que viajan hasta el ribosoma, donde el ARNm se utiliza como plantilla para la construcción de la cadena de proteínas. Dado que los ácidos nucleicos pueden unirse a moléculas con secuencias complementarias , existe una distinción entre secuencias " sentido " que codifican proteínas y la secuencia complementaria "antisentido" que no es funcional en sí misma, pero que puede unirse a la cadena sentido.
Determinación de secuencia [ editar ]
La secuenciación de ADN es el proceso de determinar la secuencia de nucleótidos de un fragmento de ADN dado . La secuencia del ADN de un ser vivo codifica la información necesaria para que ese ser vivo sobreviva y se reproduzca. Por lo tanto, determinar la secuencia es útil en la investigación fundamental sobre por qué y cómo viven los organismos, así como en temas aplicados. Debido a la importancia del ADN para los seres vivos, el conocimiento de una secuencia de ADN puede ser útil en prácticamente cualquier investigación biológica . Por ejemplo, en medicina se puede utilizar para identificar, diagnosticar y potencialmente desarrollar tratamientos para enfermedades genéticas . Del mismo modo, la investigación de patógenospuede conducir a tratamientos para enfermedades contagiosas. La biotecnología es una disciplina floreciente, con el potencial de muchos productos y servicios útiles.
El ARN no se secuencia directamente. En su lugar, se copia en un ADN mediante transcriptasa inversa , y luego este ADN se secuencia.
Los métodos de secuenciación actuales se basan en la capacidad discriminatoria de las ADN polimerasas y, por lo tanto, solo pueden distinguir cuatro bases. Una inosina (creada a partir de adenosina durante la edición de ARN ) se lee como una G, y la 5-metil-citosina (creada a partir de citosina por metilación del ADN ) se lee como una C. Con la tecnología actual, es difícil secuenciar pequeñas cantidades de ADN, ya que la señal es demasiado débil para medirla. Esto se supera mediante la amplificación de la reacción en cadena de la polimerasa (PCR).
Representación digital [ editar ]
Una vez que se ha obtenido una secuencia de ácido nucleico de un organismo, se almacena in silico en formato digital. Las secuencias genéticas digitales pueden almacenarse en bases de datos de secuencias , analizarse (ver Análisis de secuencias a continuación), modificarse digitalmente y usarse como plantillas para crear nuevo ADN real utilizando síntesis de genes artificiales .
Análisis de secuencia [ editar ]
Las secuencias genéticas digitales pueden analizarse utilizando las herramientas de la bioinformática para intentar determinar su función.
Pruebas genéticas [ editar ]
El ADN en el genoma de un organismo se puede analizar para diagnosticar vulnerabilidades a enfermedades hereditarias y también se puede utilizar para determinar la paternidad de un niño (padre genético) o la ascendencia de una persona . Normalmente, cada persona porta dos variaciones de cada gen , una heredada de su madre y la otra heredada de su padre. Se cree que el genoma humano contiene entre 20 000 y 25 000 genes. Además de estudiar los cromosomas a nivel de genes individuales, las pruebas genéticas en un sentido más amplio incluyen pruebas bioquímicas para la posible presencia de enfermedades genéticas., o formas mutantes de genes asociados con un mayor riesgo de desarrollar trastornos genéticos.
Las pruebas genéticas identifican cambios en los cromosomas, genes o proteínas. [6] Por lo general, las pruebas se utilizan para encontrar cambios asociados con trastornos hereditarios. Los resultados de una prueba genética pueden confirmar o descartar una posible afección genética o ayudar a determinar la probabilidad de que una persona desarrolle o transmita un trastorno genético. Actualmente se utilizan varios cientos de pruebas genéticas y se están desarrollando más. [7] [8]
Alineación de secuencia [ editar ]
En bioinformática, una alineación de secuencias es una forma de ordenar las secuencias de ADN , ARN o proteínas para identificar regiones de similitud que pueden deberse a relaciones funcionales, estructurales o evolutivas entre las secuencias. [9] Si dos secuencias en un alineamiento comparten un ancestro común, los desajustes se pueden interpretar como mutaciones puntuales y los espacios como mutaciones de inserción o deleción ( indels ) introducidas en uno o ambos linajes en el tiempo desde que divergieron entre sí. En las alineaciones de secuencias de proteínas, el grado de similitud entre los aminoácidosocupar una posición particular en la secuencia se puede interpretar como una medida aproximada de cuán conservada está una región particular o un motivo de secuencia entre los linajes. La ausencia de sustituciones, o la presencia de sustituciones muy conservadoras (es decir, la sustitución de aminoácidos cuyas cadenas laterales tienen propiedades bioquímicas similares) en una región particular de la secuencia, sugiere [10] que esta región tiene importancia estructural o funcional. . Aunque las bases de nucleótidos de ADN y ARN son más similares entre sí que los aminoácidos, la conservación de los pares de bases puede indicar un papel funcional o estructural similar. [11]
La filogenética computacional hace un uso extensivo de alineamientos de secuencia en la construcción e interpretación de árboles filogenéticos , que se utilizan para clasificar las relaciones evolutivas entre genes homólogos representados en los genomas de especies divergentes. El grado en que las secuencias en un conjunto de consultas difieren está cualitativamente relacionado con la distancia evolutiva de las secuencias entre sí. En términos generales, la identidad de secuencia alta sugiere que las secuencias en cuestión tienen un ancestro común más reciente comparativamente joven , mientras que la identidad baja sugiere que la divergencia es más antigua. Esta aproximación, que refleja la hipótesis del " reloj molecular " de que una tasa aproximadamente constante de cambio evolutivose puede utilizar para extrapolar el tiempo transcurrido desde que dos genes divergieron por primera vez (es decir, el tiempo de coalescencia ), se supone que los efectos de la mutación y la selección son constantes en todos los linajes de secuencia. Por lo tanto, no tiene en cuenta la posible diferencia entre organismos o especies en las tasas de reparación del ADN o la posible conservación funcional de regiones específicas en una secuencia. (En el caso de las secuencias de nucleótidos, la hipótesis del reloj molecular en su forma más básica también descuenta la diferencia en las tasas de aceptación entre mutaciones silenciosas que no alteran el significado de un codón dado y otras mutaciones que dan como resultado un aminoácido diferente incorporarse a la proteína). Los métodos estadísticamente más precisos permiten que varíe la tasa de evolución en cada rama del árbol filogenético, produciendo así mejores estimaciones de los tiempos de coalescencia de los genes.
Motivos de secuencia [ editar ]
Con frecuencia, la estructura primaria codifica motivos que son de importancia funcional. Algunos ejemplos de motivos de secuencia son: las cajas C / D [12] y H / ACA [13] de snoRNA , el sitio de unión Sm que se encuentra en los RNA espliceosomales como U1 , U2 , U4 , U5 , U6 , U12 y U3 , el Shine -Secuencia de Dalgarno , [14] la secuencia de consenso de Kozak [15] y el terminador de la ARN polimerasa III . [dieciséis]
Correlaciones de largo alcance [ editar ]
Peng y col. [17] [18] encontraron la existencia de correlaciones de largo alcance en las secuencias de pares de bases no codificantes del ADN. Por el contrario, tales correlaciones parecen no aparecer en la codificación de secuencias de ADN. Este hallazgo ha sido explicado por Grosberg et al. [19] por la estructura espacial global del ADN.
Entropía de secuencia [ editar ]
En bioinformática , una entropía de secuencia, también conocida como complejidad de secuencia o perfil de información, [20] es una secuencia numérica que proporciona una medida cuantitativa de la complejidad local de una secuencia de ADN, independientemente de la dirección de procesamiento. Las manipulaciones de los perfiles de información permiten el análisis de las secuencias utilizando técnicas sin alineación, como por ejemplo en la detección de motivos y reordenamientos. [20] [21] [22]
Ver también [ editar ]
- Estructura genética
- Sistema de numeración cuaternario
- Polimorfismo de un solo nucleótido (SNP)
Referencias [ editar ]
- ^ a b Nomenclatura para bases incompletamente especificadas en secuencias de ácidos nucleicos , NC-IUB, 1984.
- ^ Comité de nomenclatura de la Unión Internacional de Bioquímica (NC-IUB) (1984). "Nomenclatura para bases incompletamente especificadas en secuencias de ácidos nucleicos" . Consultado el 4 de febrero de 2008 .
- ^ "BIOL2060: traducción" . mun.ca .
- ^ "Investigación" . uw.edu.pl .
- ^ Nguyen, T; Brunson, D; Crespi, CL; Penman, BW; Wishnok, JS; Tannenbaum, SR (abril de 1992). "Daño y mutación del ADN en células humanas expuestas al óxido nítrico in vitro" . Proc Natl Acad Sci USA . 89 (7): 3030–034. Código Bibliográfico : 1992PNAS ... 89.3030N . doi : 10.1073 / pnas.89.7.3030 . PMC 48797 . PMID 1557408 .
- ^ "¿Qué son las pruebas genéticas?" . Referencia casera de la genética . 16 de marzo de 2015. Archivado desde el original el 29 de mayo de 2006 . Consultado el 19 de mayo de 2010 .
- ^ "Pruebas genéticas" . nih.gov .
- ^ "Definiciones de pruebas genéticas" . Definiciones de pruebas genéticas (Jorge Sequeiros y Bárbara Guimarães) . Proyecto Red de Excelencia EuroGentest. 2008-09-11. Archivado desde el original el 4 de febrero de 2009 . Consultado el 10 de agosto de 2008 .
- ^ Monte DM. (2004). Bioinformática: análisis de secuencia y genoma (2ª ed.). Prensa de laboratorio de Cold Spring Harbor: Cold Spring Harbor, NY. ISBN 0-87969-608-7.
- ^ Ng, PC; Henikoff, S. (2001). "Predecir sustituciones de aminoácidos perjudiciales" . Investigación del genoma . 11 (5): 863–74. doi : 10.1101 / gr.176601 . PMC 311071 . PMID 11337480 .
- ↑ Witzany, G (2016). "Pasos cruciales para la vida: de las reacciones químicas al código usando agentes" . Biosistemas . 140 : 49–57. doi : 10.1016 / j.biosystems.2015.12.007 . PMID 26723230 .
- ^ Samarsky, DA; Fournier MJ; Cantante RH; Bertrand E (1998). "El motivo de la caja de snoRNA C / D dirige la focalización nucleolar y también acopla la síntesis y localización de snoRNA" . El diario EMBO . 17 (13): 3747–57. doi : 10.1093 / emboj / 17.13.3747 . PMC 1170710 . PMID 9649444 .
- ^ Ganot, Philippe; Caizergues-Ferrer, Michèle; Kiss, Tamás (1 de abril de 1997). "La familia de ARN nucleolares pequeños de caja ACA se define por una estructura secundaria conservada evolutivamente y elementos de secuencia ubicuos esenciales para la acumulación de ARN" . Genes y desarrollo . 11 (7): 941–56. doi : 10.1101 / gad.11.7.941 . PMID 9106664 .
- ^ Shine J, Dalgarno L (1975). "Determinante de la especificidad del cistrón en los ribosomas bacterianos". Naturaleza . 254 (5495): 34–38. Código Bibliográfico : 1975Natur.254 ... 34S . doi : 10.1038 / 254034a0 . PMID 803646 . S2CID 4162567 .
- ^ Kozak M (octubre de 1987). "Un análisis de secuencias no codificantes 5 'de 699 ARN mensajeros de vertebrados" . Ácidos nucleicos Res . 15 (20): 8125–48. doi : 10.1093 / nar / 15.20.8125 . PMC 306349 . PMID 3313277 .
- ^ Bogenhagen DF, Brown DD (1981). "Secuencias de nucleótidos en el ADN de Xenopus 5S necesarias para la terminación de la transcripción". Celular . 24 (1): 261–70. doi : 10.1016 / 0092-8674 (81) 90522-5 . PMID 6263489 . S2CID 9982829 .
- ^ Peng, C.-K .; Buldyrev, SV; Goldberger, AL; Havlin, S .; Sciortino, F .; Simons, M .; Stanley, HE (1992). "Correlaciones de largo alcance en secuencias de nucleótidos". Naturaleza . 356 (6365): 168–70. Código Bibliográfico : 1992Natur.356..168P . doi : 10.1038 / 356168a0 . ISSN 0028-0836 . PMID 1301010 . S2CID 4334674 .
- ^ Peng, C.-K .; Buldyrev, SV; Havlin, S .; Simons, M .; Stanley, HE; Goldberger, AL (1994). "Organización en mosaico de nucleótidos del ADN" . Revisión E física . 49 (2): 1685–89. Código Bibliográfico : 1994PhRvE..49.1685P . doi : 10.1103 / PhysRevE.49.1685 . ISSN 1063-651X . PMID 9961383 .
- ^ Grosberg, A; Rabin, Y; Havlin, S; Neer, A (1993). "Modelo de glóbulo arrugado de la estructura tridimensional del ADN". Cartas de Europhysics . 23 (5): 373–78. Código Bibliográfico : 1993EL ..... 23..373G . doi : 10.1209 / 0295-5075 / 23/5/012 .
- ^ a b Pinho, A; García, S; Pratas, D; Ferreira, P (21 de noviembre de 2013). "Secuencias de ADN de un vistazo" . PLOS ONE . 8 (11): e79922. Código Bibliográfico : 2013PLoSO ... 879922P . doi : 10.1371 / journal.pone.0079922 . PMC 3836782 . PMID 24278218 .
- ^ Pratas, D; Silva, R; Pinho, A; Ferreira, P (18 de mayo de 2015). "Un método sin alineación para encontrar y visualizar reordenamientos entre pares de secuencias de ADN" . Informes científicos . 5 : 10203. Código Bibliográfico : 2015NatSR ... 510203P . doi : 10.1038 / srep10203 . PMC 4434998 . PMID 25984837 .
- ↑ Troyanskaya, O; Arbell, O; Koren, Y; Landau, G; Bolshoy, A (2002). "Perfiles de complejidad de secuencia de secuencias genómicas procariotas: un algoritmo rápido para calcular la complejidad lingüística" . Bioinformática . 18 (5): 679–88. doi : 10.1093 / bioinformatics / 18.5.679 . PMID 12050064 .
Enlaces externos [ editar ]
| Wikimedia Commons tiene medios relacionados con la secuencia del ácido nucleico . |
- Una bibliografía sobre características, patrones, correlaciones en textos de ADN y proteínas.