| Cicloadición 1,3-dipolar de Huisgen | |
|---|---|
| Lleva el nombre de | Rolf Huisgen |
| Tipo de reacción | Reacción de formación de anillo |
| Identificadores | |
| Portal de química orgánica | huisgen-1,3-cicloadición-dipolar |
| ID de ontología RSC | RXNO: 0000018 |
La cicloadición 1,3-dipolar es una reacción química entre un 1,3-dipolo y un dipolarófilo para formar un anillo de cinco miembros. Las primeras cicloadiciones 1,3-dipolares se describieron a finales del siglo XIX y principios del siglo XX, tras el descubrimiento de los 1,3-dipolos. La investigación mecanicista y la aplicación sintética se establecieron en la década de 1960, principalmente a través del trabajo de Rolf Huisgen . [1] Por lo tanto, la reacción a veces se denomina cicloadición de Huisgen (este término se usa a menudo para describir específicamente la cicloadición 1,3-dipolar entre unazida y un alquino para generar 1,2,3-triazol ). La cicloadición 1,3-dipolar es una ruta importante para la síntesis regio y estereoselectiva de heterociclos de cinco miembros y sus derivados acíclicos de anillo abierto. El dipololarófilo es típicamente un alqueno o alquino, pero puede ser otros sistemas pi. Cuando el dipololarófilo es un alquino, generalmente se producen anillos aromáticos.

Resumen mecanicista
Originalmente, dos mecanismos propuestos describen la cicloadición 1,3-dipolar: primero, el mecanismo de cicloadición pericíclica concertada , propuesto por Rolf Huisgen; [2] y segundo, el mecanismo escalonado que involucra un intermedio dirradical , propuesto por Firestone. [3] Después de mucho debate, la primera propuesta ahora es generalmente aceptada [4] - el 1,3-dipolo reacciona con el dipolo en una forma concertada , a menudo asincrónica y simetría -permitida π 4 s + π 2 s a través de una Estado de transición aromático Huckel de seis electrones . Sin embargo, existen algunos ejemplos de un mecanismo escalonado para las reacciones de cicloadición 1,3-dipolar sin catalizador de iluros de tiocarbonilo, [5] y óxidos de nitrilo [6].

Mecanismo pericíclico
Huisgen investigó una serie de cicloadiciones entre los compuestos diazo 1,3-dipolares y varios alquenos dipolarofílicos . [2] Las siguientes observaciones apoyan el mecanismo pericíclico concertado y refutan la vía direccional escalonada o polar escalonada.
- Efectos de los sustituyentes : diferentes sustituyentes en el dipolo no muestran un gran efecto sobre la velocidad de cicloadición, lo que sugiere que la reacción no implica un intermedio de carga separada.
- Efectos del solvente : La polaridad del solvente tiene poco efecto sobre la tasa de cicloadición, en línea con el mecanismo pericíclico donde la polaridad no cambia mucho al pasar de los reactivos al estado de transición.
- Estereoquímica : las cicloadiciones 1,3-dipolares son siempre estereoespecíficas con respecto al dipolarófilo (es decir, cis -alquenos que dan productos syn ), lo que respalda el mecanismo pericíclico concertado en el quese formandos enlaces sigma simultáneamente.
- Parámetros termodinámicos : las cicloadiciones 1,3-dipolares tienen una entropía de activación negativa inusualmente grandesimilar a la de la reacción de Diels-Alder , lo que sugiere que el estado de transición está muy ordenado, que es una firma de reacciones pericíclicas concertadas.
1,3-dipolo
Un 1,3-dipolo es una molécula orgánica que puede representarse como un tipo alilo o una estructura de sexteto / octeto zwiteriónico de tipo propargilo / alenilo . Ambos tipos de 1,3-dipolos comparten cuatro electrones en el sistema π sobre tres átomos. El tipo alilo está doblado mientras que el tipo propargilo / alenilo es lineal en geometría . [7] También se conocen 1,3-dipolos que contienen elementos de filas superiores como azufre o fósforo , pero se utilizan de forma menos rutinaria.
Se pueden dibujar estructuras de resonancia para deslocalizar tanto las cargas negativas como las positivas en cualquier terminal de un 1,3-dipolo (ver el esquema a continuación). Un método más preciso para describir la distribución electrónica en un 1,3-dipolo es asignar el principal contribuyente de resonancia en base a datos experimentales o teóricos, como mediciones de momento dipolar [8] o cálculos. [9] Por ejemplo, el diazometano tiene el carácter negativo más grande en el átomo de nitrógeno terminal , mientras que el ácido hidrazoico tiene el carácter negativo más grande en el átomo de nitrógeno interno .
En consecuencia, esta ambivalencia significa que los extremos de un 1,3-dipolo pueden tratarse como nucleófilos y electrófilos al mismo tiempo. El grado de nucleofilia y electrofilia en cada extremo se puede evaluar utilizando los orbitales moleculares de frontera , que se pueden obtener computacionalmente. En general, el átomo que lleva el coeficiente orbital más grande en el HOMO actúa como nucleófilo, mientras que en el LUMO actúa como electrófilo. El átomo más nucleofílico suele ser, pero no siempre, el átomo más rico en electrones. [10] [11] [12] En las cicloadiciones 1,3-dipolares, la identidad del par dipolo-dipolarófilo determina si dominará el carácter HOMO o LUMO del 1,3-dipolo (véase más adelante la discusión sobre los orbitales moleculares de frontera).
Dipolófilo
Los dipolarófilos más utilizados son los alquenos y alquinos. Los dipolos que contienen heteroátomos , como los carbonilos y las iminas , también pueden sufrir cicloadición 1,3-dipolar. Otros ejemplos de dipolarófilos incluyen fullerenos y nanotubos , que pueden sufrir cicloadición 1,3-dipolar con iluro de azometina en la reacción de Prato .
Efectos solventes
Las cicloadiciones 1,3-dipolares experimentan muy poco efecto disolvente porque tanto los reactivos como los estados de transición son generalmente no polares. Por ejemplo, la velocidad de reacción entre el fenil diazometano y el acrilato de etilo o el norborneno (véase el esquema a continuación) cambia solo ligeramente al variar los disolventes desde el ciclohexano hasta el metanol. [13]
La falta de efectos de los disolventes en la cicloadición 1,3-dipolar se demuestra claramente en la reacción entre las enaminas y el dimetil diazomalonato (ver esquema a continuación). [14] La reacción polar, N-ciclo pluma tenyl pirrolidina adición nucleófila al compuesto diazo, procede 1.500 veces más rápido en polar DMSO que en no polar decalina . Por otro lado, un cierre analógica de esta reacción, N-ciclo hex enil pirrolidina 1,3-dipolar de cicloadición de dimetil diazomalonato, se acelera solamente 41 veces en DMSO en relación con decalina.
Teoría de los orbitales moleculares de frontera
Las cicloadiciones 1,3-dipolares son reacciones pericíclicas, que obedecen las reglas de Dewar-Zimmerman y las reglas de Woodward-Hoffmann . En el tratamiento de Dewar-Zimmerman, la reacción avanza a través de un estado de transición de Huckel de 5 centros, cero nodos y 6 electrones para este diagrama de orbitales moleculares en particular. Sin embargo, a cada orbital se le puede asignar un signo al azar para llegar al mismo resultado. En el tratamiento de Woodward-Hoffmann, los orbitales moleculares de frontera (FMO) del 1,3-dipolo y el dipololarófilo se superponen de la manera π 4 s + π 2 s permitida por la simetría . Tal superposición orbital se puede lograr de tres formas: tipo I, II y III. [15] La vía dominante es la que posee la brecha de energía HOMO-LUMO más pequeña.
Tipo I
El dipolo tiene un HOMO elevado que se superpone con el LUMO del dipolo . Un dipolo de esta clase se denomina dipolo controlado por HOMO o dipolo nucleofílico , que incluye iluro de azometina , iluro de carbonilo , iluro de nitrilo , imina de azometina , imina de carbonilo y diazoalcano.. Estos dipolos se agregan fácilmente a los alquenos electrofílicos. Los grupos sustractores de electrones (EWG) en el dipololarófilo acelerarían la reacción al disminuir el LUMO, mientras que los grupos donadores de electrones (EDG) desacelerarían la reacción al aumentar el HOMO. Por ejemplo, en el siguiente esquema se muestra la escala de reactividad del diazometano frente a una serie de dipolos. El diazometano reacciona con el acrilato de etilo pobre en electrones más de un millón de veces más rápido que el éter butilvinílico rico en electrones. [dieciséis]
Este tipo se asemeja a la reacción de Diels-Alder de demanda normal de electrones, en la que el dieno HOMO se combina con el dienófilo LUMO.
Tipo II
HOMO del dipolo puede emparejarse con LUMO del dipolo; alternativamente, HOMO del dipolo puede emparejarse con LUMO del dipolo. Esta interacción bidireccional surge porque la brecha de energía en cualquier dirección es similar. Un dipolo de esta clase se conoce como dipolo controlado por HOMO-LUMO o dipolo ambifílico , que incluye imida de nitrilo , nitrona , óxido de carbonilo , óxido de nitrilo y azida.. Cualquier sustituyente en el dipololarófilo aceleraría la reacción al reducir la brecha de energía entre los dos orbitales que interactúan; es decir, un EWG bajaría el LUMO mientras que un EDG aumentaría el HOMO. Por ejemplo, las azidas reaccionan con varios dipolocrófilos ricos y pobres en electrones con reactividades similares (ver la escala de reactividad a continuación). [17]
Tipo III
El dipolo tiene un LUMO bajo que se superpone con el HOMO del dipolo (indicado por líneas rojas discontinuas en el diagrama). Un dipolo de esta clase se denomina dipolo controlado por LUMO o dipolo electrofílico , que incluye óxido nitroso y ozono . Los EWG en el dipololarófilo desaceleran la reacción, mientras que los EDG aceleran la reacción. Por ejemplo, el ozono reacciona con el 2-metilpropeno rico en electrones aproximadamente 100.000 veces más rápido que el tetracloroeteno pobre en electrones (ver la escala de reactividad a continuación). [18]
Este tipo se asemeja a la reacción de Diels-Alder por demanda inversa de electrones , en la que el dieno LUMO se combina con el dienophile HOMO.
Reactividad
Los procesos concertados como la 1,3-cicloadición requieren un estado de transición altamente ordenado (alta entropía negativa de activación) y solo requisitos de entalpía moderados. Utilizando experimentos de reacción competitiva, se ha encontrado que las velocidades relativas de adición para diferentes reacciones de cicloadición ofrecen hallazgos generales sobre los factores de reactividad.
- La conjugación , especialmente con grupos aromáticos, aumenta la velocidad de reacción mediante la estabilización del estado de transición. Durante la transición, los dos enlaces sigma se forman a diferentes velocidades, lo que puede generar cargas parciales en el estado de transición que pueden estabilizarse mediante la distribución de carga en sustituyentes conjugados.
- Los dipolocrófilos más polarizables son más reactivos porque las nubes de electrones difusos son más adecuadas para iniciar el flujo de electrones.
- Los dipolorófilos con alta deformación angular son más reactivos debido al aumento de energía del estado fundamental.
- El aumento del impedimento estérico en el estado de transición como resultado de los reactivos no impedidos reduce drásticamente la velocidad de reacción.
- Los heterodipolarófilos se agregan más lentamente, si es que lo hacen, en comparación con los C, C-diapolarófilos debido a una menor ganancia en la energía del enlace sigma para compensar la pérdida de un enlace pi durante el estado de transición.
- La isomería del dipolocrófilo afecta la velocidad de reacción debido a los estéricos. Los isómeros trans son más reactivos (el trans -stilbeno agregará difenil (nitrilo imida) 27 veces más rápido que el cis -stilbeno) porque durante la reacción, el ángulo de enlace de 120 ° se contrae a 109 °, llevando los sustituyentes cis eclipsantes entre sí para aumentar choque estérico.

Estereoespecificidad
Las cicloadiciones 1,3-dipolares normalmente dan como resultado la retención de la configuración con respecto tanto al 1,3-dipolo como al dipolo- rófilo. Este alto grado de estereoespecificidad es un fuerte apoyo para los mecanismos de reacción concertados sobre los escalonados. Como se mencionó anteriormente, muchos ejemplos muestran que las reacciones fueron escalonadas, presentando así una estereoespecificidad parcial o nula.
Con respecto al dipololarófilo
Los sustituyentes cis en el alqueno dipolarofílico terminan en cis , y los sustituyentes trans terminan en trans en el compuesto cíclico de cinco miembros resultante (ver esquema a continuación). [19]
Con respecto al dipolo
Generalmente, la estereoquímica del dipolo no es de gran importancia porque solo unos pocos dipolos podrían formar centros estereogénicos , y las estructuras de resonancia permiten la rotación de enlaces que codifica la estereoquímica. Sin embargo, el estudio de iluros de azometina ha verificado que la cicloadición también es estereoespecífica con respecto al componente dipolo. Los iluros de azometina diastereopuros se generan mediante la apertura del anillo electrocíclico de aziridinas , y luego se atrapan rápidamente con dipolarófilos fuertes antes de que pueda tener lugar la rotación del enlace (ver esquema a continuación). [20] [21] Si se utilizan dipolos más débiles, los enlaces en el dipolo tienen la posibilidad de rotar, lo que da como resultado una estereoespecificidad de cicloadición deteriorada.
Estos resultados confirman por completo que la cicloadición 1,3-dipolar es estereoespecífica, lo que da retención tanto del 1,3-dipolo como del dipololarófilo.
Diastereoselectividad
Cuando se generan dos o más estereocentros durante la reacción, se pueden obtener estados y productos de transición diastereoméricos. En la cicloadición de Diels-Alder, se suele observar la endo diastereoselectividad debida a interacciones orbitales secundarias . En las cicloadiciones 1,3-dipolares, sin embargo, dos fuerzas influyen en la diastereoselectividad: la interacción π atractiva (que se asemeja a las interacciones orbitales secundarias en la cicloadición de Diels-Alder) y la interacción estérica repulsiva . Desafortunadamente, estas dos fuerzas a menudo se cancelan entre sí, provocando una mala diastereoselección en la cicloadición 1,3-dipolar.
A continuación se muestran ejemplos de cicloadiciones 1,3-dipolares diastereoselectivas controladas por sustrato. Primero está la reacción entre el benzonitrilo N-benciluro y el acrilato de metilo . En el estado de transición, los grupos fenilo y éster metílico se apilan para dar la sustitución cis como el producto de pirrolina final exclusivo . Esta interacción π favorable compensa la repulsión estérica entre los grupos fenilo y éster metílico. [22] El segundo es la reacción entre la nitrona y el dihidrofurano . La exo- selectividad se logra para minimizar la repulsión estérica. [23]Por último, está la reacción de iluro de azometina intramolecular con alqueno. La diastereoselectividad se controla mediante la formación de un menos tensas cis - sistema de anillos condensados . [24]
Cicloadición 1,3-dipolar dirigida
La trayectoria de la cicloadición se puede controlar para lograr una reacción diastereoselectiva. Por ejemplo, los metales pueden quelarse al dipolo y al dipolo entrante y dirigir la cicloadición selectivamente en una cara. El siguiente ejemplo muestra la adición de óxido de nitrilo a un alcohol alílico enantioméricamente puro en presencia de un ion magnesio. La conformación más estable del alqueno coloca al grupo hidroxilo por encima del plano del alqueno. Luego, el magnesio se quela al grupo hidroxilo y al átomo de oxígeno del óxido de nitrilo. Por tanto, la cicloadición procede de la cara superior de forma selectiva. [25]
Tal diastereodirección se ha aplicado en la síntesis de epotilonas . [26]
Regioselectividad
Para pares asimétricos dipolo-dipololarófilos, son posibles dos productos regioisoméricos . Tanto los factores electrónicos / estereoelectrónicos como los estéricos contribuyen a la regioselectividad de las cicloadiciones 1,3-dipolares. [27]
Efecto electrónico / estereoelectrónico
La interacción electrónica dominante es la combinación entre el HOMO más grande y el LUMO más grande. Por lo tanto, la regioselectividad está gobernada por los átomos que portan los coeficientes orbitales HOMO y LUMO más grandes. [28] [29]
Por ejemplo, considérese la cicloadición de diazometano a tres dipolarófilos: acrilato de metilo , estireno o cinamato de metilo . El carbono del diazometano tiene el mayor HOMO, mientras que los carbonos olefínicos finales del acrilato de metilo y el estireno tienen el mayor LUMO. Por tanto, la cicloadición da la sustitución en la posición C-3 de forma regioselectiva. Para el cinamato de metilo, los dos sustituyentes (Ph vs COOMe) compiten en la extracción de electrones del alqueno. El carboxilo es el mejor grupo captador de electrones, lo que hace que el carbono β sea más electrófilo. Por tanto, la cicloadición produce el grupo carboxilo en C-3 y el grupo fenilo en C-4 regioselectivamente.
Efecto estérico
Los efectos estéricos pueden cooperar o competir con los efectos electrónicos antes mencionados. A veces, los efectos estéricos superan por completo la preferencia electrónica, dando exclusivamente el regioisómero opuesto. [30]
Por ejemplo, el diazometano generalmente se agrega al acrilato de metilo para dar 3-carboxil pirazolina . Sin embargo, al poner más demandas estéricas en el sistema, comenzamos a observar las 4-carboxil pirazolinas isoméricas. La proporción de estos dos regioisómeros depende de las demandas estéricas. En el extremo, aumentar el tamaño de hidrógeno a t-butilo desplaza la regioselectividad del 100% de sustitución de 3-carboxilo al 100% de sustitución de 4-carboxilo. [31] [32]
Aplicaciones sintéticas
Las cicloadiciones 1,3-dipolares son formas importantes de síntesis de muchos heterociclos importantes de 5 miembros, como triazoles , furanos , isoxazoles , pirrolidinas y otros. Además, algunos cycloadducts se pueden escindir para revelar el esqueleto lineal, proporcionando otra ruta hacia la síntesis de compuestos alifáticos . Estas reacciones son tremendamente útiles también porque son estereoespecíficas, diastereoselectivas y regioselectivas. A continuación se proporcionan varios ejemplos.
Óxidos de nitrilo
La cicloadición 1,3-dipolar con óxidos de nitrilo es una reacción aldólica enmascarada muy utilizada . La cicloadición entre un óxido de nitrilo y un alqueno produce el producto de isoxazolina cíclico, mientras que la reacción con un alquino produce el isoxazol. Tanto las isoxazolinas como los isoxazoles pueden escindirse por hidrogenación para revelar productos β-hidroxicarbonilo de tipo aldol o β-dicarbonilo de tipo Claisen , respectivamente.
En la síntesis de Miyakolide se utilizó cicloadición de óxido de nitrilo-alquino seguida de hidrogenación como se ilustra en la figura siguiente. [33]
Iluros de carbonilo
Las reacciones de cicloadición 1,3-dipolar han surgido como herramientas poderosas en la síntesis de andamios y moléculas cíclicas complejas para estudios medicinales, biológicos y mecanicistas. Entre ellos, las reacciones de cicloadición [3 + 2] que involucran iluros de carbonilo se han empleado ampliamente para generar moléculas cíclicas de cinco miembros que contienen oxígeno. [34]
Preparación de iluros de carbonilo para reacciones de cicloadición 1,3-dipolar
Los iluros se consideran heteroátomos cargados positivamente conectados a átomos de carbono cargados negativamente, que incluyen iluros de sulfonio , tiocarbonilo , oxonio , nitrógeno y carbonilo . [35] Existen varios métodos para generar iluros de carbonilo, que son intermedios necesarios para generar estructuras de anillo de cinco miembros que contienen oxígeno, para reacciones de cicloadición [3 + 2].
Síntesis de iluros de carbonilo a partir de derivados de diazometano por fotocatálisis
Uno de los primeros ejemplos de síntesis de iluro de carbonilo implica la fotocatálisis . [36] La fotólisis de diazotetrakis (trifluorometil) ciclopentadieno * (DTTC) en presencia de tetrametilurea puede generar el iluro de carbonilo mediante un ataque nucleófilo intermolecular y la posterior aromatización del resto DTTC. [36] Esto se aisló y caracterizó mediante cristalografía de rayos X debido a la estabilidad impartida por la aromaticidad, los grupos trifluorometilo que extraen electrones y los grupos dimetilamina que dan electrones . Dipolos de iluro de carbonilo establesluego se puede utilizar en reacciones de cicloadición [3 + 2] con dipololarófilos .

Otro ejemplo temprano de síntesis de iluro de carbonilo por fotocatálisis fue informado por Olah et al . [37] El dideuteriodiazometano se fotolizó en presencia de formaldehído para generar el dideuterioformaldehído carbonil iluro.

Síntesis de iluros de carbonilo a partir de hidroxipironas por transferencia de protones
Los iluros de carbonilo se pueden sintetizar mediante catálisis ácida de hidroxi-3-pironas en ausencia de un catalizador metálico . [38] Se produce una tautomerización inicial , seguida de la eliminación del grupo saliente para aromatizar el anillo de pirona y generar el iluro de carbonilo. Una reacción de cicloadición con un dipolarófilo forma, por último, la oxacycle. Este enfoque se emplea menos ampliamente debido a su utilidad y requisito limitados para esqueletos de pirona.

También se pueden usar 5-hidroxi-4-pironas para sintetizar iluros de carbonilo mediante una transferencia de hidrógeno intramolecular . [39] Después de la transferencia de hidrógeno, el iluro de carbonilo puede reaccionar con dipolos para formar anillos que contienen oxígeno.

Síntesis de iluros de α-halocarbonilo a partir de dihalocarbenos
También se han empleado dihalocarbenos para generar iluros de carbonilo, aprovechando la naturaleza aceptora de electrones de los dihalocarbenos. [40] [41] [42] Tanto el fenil (triclorometil) mercurio como el fenil (tribromometil) mercurio son fuentes de diclorocarbenos y dibromocarbenos , respectivamente. El iluro de carbonilo se puede generar tras la reacción de los dihalocarbenos con cetonas o aldehídos . Sin embargo, la síntesis de iluros de α-halocarbonilo también puede conducir indeseablemente a la pérdida de monóxido de carbono y la generación del producto de desoxigenación.

Síntesis de iluros de carbonilo a partir de derivados de diazometano mediante catálisis de metales
Un enfoque universal para generar iluros de carbonilo implica la catálisis de metales de compuestos de α-diazocarbonilo, generalmente en presencia de catalizadores de dicopper o dirhodium. [43] Después de la liberación de nitrógeno gaseoso y la conversión en metalocarbeno , una reacción intermolecular con un grupo carbonilo puede generar el iluro de carbonilo. Reacción de cicloadición posterior con un alqueno o alquino dipolarófilo puede permitirse anillos que contienen oxígeno de cinco miembros. Los catalizadores populares que dan rendimientos modestos para sintetizar oxaciclos incluyen Rh 2 (OAc) 4 y Cu (acac) 2 . [44] [45]

Mecanismo de la reacción de cicloadición 1,3-dipolar mediada por catálisis de metales de compuestos de diazocarbonilo
La universalidad y el uso extensivo de las reacciones de cicloadición 1,3-dipolar mediadas por catálisis de metales de moléculas de diazocarbonilo, para sintetizar anillos de cinco miembros que contienen oxígeno, ha despertado un interés significativo en su mecanismo. Varios grupos han investigado el mecanismo para ampliar el alcance de las moléculas sintéticas con respecto a la regio y estereo-selectividad . Sin embargo, debido a las altas frecuencias de rotación de estas reacciones, los intermediarios y el mecanismo siguen siendo esquivos. El mecanismo generalmente aceptado, desarrollado por la caracterización de complejos estables de rutenio-carbenoide [46] y metalocarbenos de rodio, [47] implica una formación inicial de un complejo de metal-carbenoide a partir delcompuesto diazo . La eliminación de nitrógeno gaseoso produce un metalocarbeno. Un ataque nucleofílico intramolecular por el oxígeno del carbonilo regenera el catalizador metálico y forma el iluro de carbonilo. El iluro de carbonilo puede entonces reaccionar con un alqueno o alquino, como acetilendicarboxilato de dimetilo (DMAD) para generar el oxaciclo.

Sin embargo, no está claro si el intermedio de metalocarbeno genera el iluro de carbonilo. En algunos casos, los metalocarbenos también pueden reaccionar directamente con dipolos. [48] En estos casos, el metalocarbeno, como el tetracarboxilato carbeno de dirodio (II), se estabiliza mediante interacciones de tipo enolato metálico hiperconjugativo . [49] [50] [51] [52] La posterior reacción de cicloadición 1,3-dipolar se produce a través de un iluro de carbonilo complejado con un metal transitorio. Por lo tanto, un metalocarbeno persistente puede influir en la estereoselectividad y regioselectividad de la reacción de cicloadición 1,3-dipolar en función de la estereoquímica y el tamaño de los ligandos metálicos .

El mecanismo de la reacción de cicloadición 1,3-dipolar entre el dipolo de iluro de carbonilo y los dipolarófilos de alquinilo o alquenilo se ha investigado extensamente con respecto a la regioselectividad y la estereoselectividad. Como los dipolocrófilos simétricos tienen una orientación para la cicloadición, solo se puede obtener un regioisómero , pero se pueden obtener múltiples estereoisómeros . [52] Por el contrario, los dipolos asimétricos pueden tener múltiples regioisómeros y estereoisómeros. Estos regioisómeros y estereoisómeros pueden predecirse basándose en la teoría de orbitales moleculares fronterizos (FMO) , interacciones estéricas yInteracciones estereoelectrónicas . [53] [54]

Regioselectividad de la reacción de cicloadición 1,3-dipolar mediada por catálisis de metales de compuestos de diazocarbonilo
La regioselectividad de las reacciones de cicloadición 1,3-dipolar entre dipolos de iluro de carbonilo y dipolarófilos de alquinilo o alquenilo es esencial para generar moléculas con regioquímica definida. La teoría FMO y el análisis de las brechas de energía HOMO-LUMO entre el dipolo y el dipololarófilo pueden racionalizar y predecir la regioselectividad de los resultados experimentales. [55] [56] Los HOMOs y lumos pueden pertenecer a cualquiera de los dos el dipolo o dipolarófilo, para lo cual HOMO dipolo -LUMO dipolarófilo o HOMO dipolarófilo -LUMO dipolares pueden existir interacciones. La superposición de los orbitales con los coeficientes más grandes puede, en última instancia, racionalizar y predecir los resultados.

Padwa y colaboradores examinaron la regioselectividad arquetípica de la reacción de cicloadición 1,3-dipolar mediada por dipolos de iluro de carbonilo. [54] [57] El uso de un Rh 2 (OAc) 4 catalizador en benceno, diazodione se sometió a una reacción de cicloadición 1,3-dipolar con metil propiolato de metilo y propargilo éter . La reacción con propiolato de metilo proporciona dos regioisómeros con la mayor resultante de la HOMO dipolo -LUMO dipolarófilointeracción, que tiene los coeficientes más grandes en el carbono próximo al grupo carbonilo del iluro de carbonilo y en el carbono alquino terminal propiolato de metilo. La reacción con metil propargil éter produce un regioisómero resultante de la interacción dipolarófilo - dipolo LUMO de HOMO , que tiene los mayores coeficientes en el carbono distal al grupo carbonilo del iluro de carbonilo y en el carbono alquino terminal del metil propargil éter.
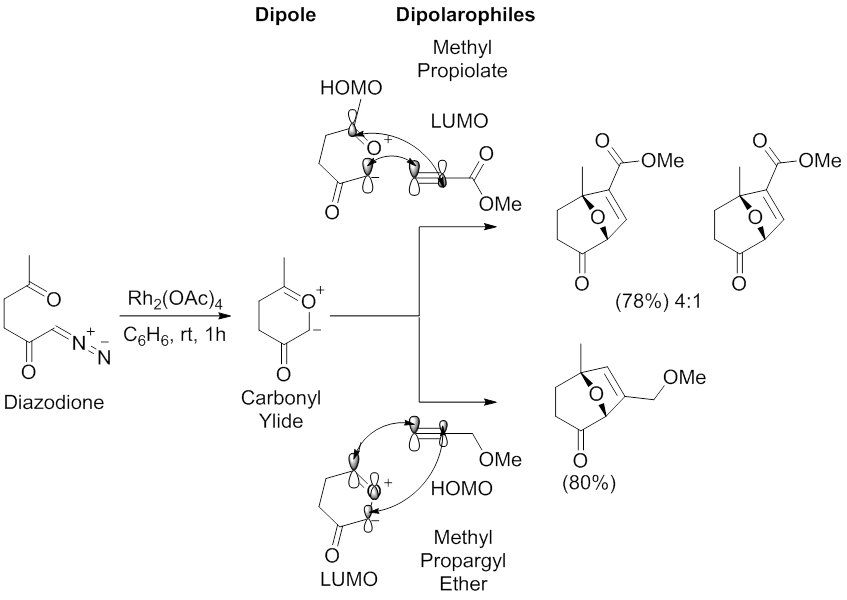
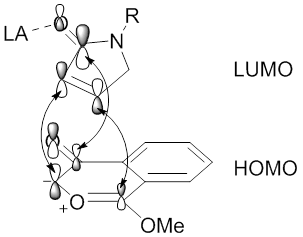
Las regioselectividades de las reacciones de cicloadición 1,3-dipolar mediadas por catálisis de metales de compuestos de diazocarbonilo también pueden estar influenciadas por el metal a través de la formación de metalocarbenos estables. [48] [58] La estabilización del metalocarbeno, a través de interacciones de tipo enolato de metal, evitará la formación de iluros de carbonilo, lo que dará como resultado una reacción directa entre el dipolo de metalocarbeno y un alquinilo o alquenil dipolo (ver imagen del dirodio (II) metalocarbeno tetracarboxilato estabilizado por hiperconjugación π C-Rh → π C = O ). En esta situación, los ligandos metálicos influirán en la regioselectividad y la estereoselectividad de la reacción de cicloadición 1,3-dipolar.
Estereoselectividad e inducción asimétrica de la reacción de cicloadición 1,3-dipolar mediada por catálisis de metales de compuestos de diazocarbonilo
La estereoselectividad de las reacciones de cicloadición 1,3-dipolar entre dipolos iluro carbonilo y dipolarófilos alquenilo también se ha examinado de cerca. Para los alquinil dipolarófilos, la estereoselectividad no es un problema ya que se forman carbonos sp 2 relativamente planos , mientras que se debe considerar la regioselectividad (ver imagen de los productos de la reacción de cicloadición 1,3-dipolar entre dipolos de iluro de carbonilo y dipolos de alquenilo o alquinilo). Sin embargo, para los alquenildipolarófilos, se deben considerar tanto la regioselectividad como la estereoselectividad, ya que se generan carbonos sp 3 en la especie del producto.
Las reacciones de cicloadición 1,3-dipolar entre dipolos de iluro de carbonilo y dipolarófilos de alquenilo pueden generar productos diastereoméricos . [52] El producto exo se caracteriza con sustituyentes dipolarófilos que son cis al puente éter del oxaciclo. El producto endo se caracteriza por ser los sustituyentes dipolorófilos trans al puente de éter del oxaciclo. Ambos productos pueden ser generados a través de pericíclicas transiciones estados que implican concertadas procesos asíncronos síncronos o concertadas.
Un ejemplo temprano confirió estereoselectividad en términos de productos endo y exo con catalizadores metálicos y ácidos de Lewis. [59] Las reacciones sólo con el catalizador metálico Rh 2 (OAc) 4 prefieren el producto exo , mientras que las reacciones con el ácido de Lewis adicional Yb (OTf) 3 prefieren el producto endo . La endo selectividad observada para las reacciones de cicloadición ácida de Lewis se atribuye a la superposición orbital optimizada de los sistemas de carbonilo π entre el dipolarófilo coordinado por Yb (Otf) 3(LUMO) y el dipolo (HOMO). Después de muchas investigaciones, se han desarrollado dos enfoques principales para influir en la estereoselectividad de las cicloadiciones de iluro de carbonilo que explotan la quiralidad de los catalizadores metálicos y los ácidos de Lewis. [52]

El primer enfoque emplea catalizadores de metales quirales para modular la estereoselectividad endo y exo . Los catalizadores quirales, en particular Rh 2 [( S ) -DOSP] 4 y Rh 2 [( S ) -BPTV] 4 pueden inducir una inducción asimétrica modesta y se usaron para sintetizar el agente antifúngico ácido pseudolarico A. [60] Este es un resultado de que el catalizador de metal quiral permanece asociado con el iluro de carbonilo durante la cicloadición, lo que confiere selectividad facial. Sin embargo, los mecanismos exactos aún no se comprenden completamente.

El segundo enfoque emplea un catalizador de ácido de Lewis quiral para inducir la estereoselectividad facial después de la generación del iluro de carbonilo usando un catalizador metálico aquiral. [61] Se cree que el catalizador de ácido de Lewis quiral se coordina con el dipololarófilo, lo que reduce el LUMO del diplarófilo al tiempo que conduce a la enantioselectividad .

Iluros de azometina
La cicloadición 1,3-dipolar entre un iluro de azometina y un alqueno proporciona una estructura azacíclica, como la pirrolidina . Esta estrategia se ha aplicado a la síntesis de espirotriprostatina A. [62]
Ozono
La ozonólisis es una reacción orgánica muy importante. Los alquenos y alquinos pueden escindirse mediante ozonólisis para dar productos de aldehído , cetona o ácido carboxílico .
Aplicaciones biológicas
La cicloadición 1,3-dipolar entre azidas orgánicas y alquinos terminales (es decir, la cicloadición de Huisgen ) se ha utilizado ampliamente para la bioconjugación .
Catálisis de cobre
La reacción de Huisgen generalmente no se desarrolla fácilmente en condiciones suaves. Meldal y col. y Sharpless et al. desarrolló de forma independiente una versión catalizada por cobre (I) de la reacción de Huisgen, CuAAC (para cicloadición azida-alquino catalizada por cobre), que avanza muy fácilmente en condiciones suaves, incluidas las fisiológicas ( pH neutro , temperatura ambiente y solución acuosa ). [63] [64] Esta reacción también es bioortogonal : las azidas y los alquinos generalmente están ausentes de los sistemas biológicos y, por lo tanto, estas funcionalidades pueden reaccionar quimioselectivamente incluso en elcontexto celular . Tampoco reaccionan con otros grupos funcionales que se encuentran en la naturaleza, por lo que no perturban los sistemas biológicos. La reacción es tan versátil que se denomina química del "clic" . Aunque el cobre (I) es tóxico , se han desarrollado muchos ligandos protectores para reducir la citotoxicidad y mejorar la tasa de CuAAC, lo que permite su uso en estudios in vivo . [sesenta y cinco]
Por ejemplo, Bertozzi et al. informaron de la incorporación metabólica de sacáridos funcionalizados con azida en el glicano de la membrana celular y el marcado posterior con conjugado de fluoróforo- alquino. La membrana celular ahora marcada con fluorescencia se puede visualizar bajo el microscopio . [66]
Cicloadición promovida por cepas
Para evitar la toxicidad del cobre (I), Bertozzi et al. desarrollaron la cicloadición azida-alquino promovida por cepa (SPAAC) entre azida orgánica y ciclooctina tensada . La distorsión del ángulo de la ciclooctina ayuda a acelerar la reacción reduciendo la tensión de activación y mejorando las interacciones, lo que permite su uso en condiciones fisiológicas sin la necesidad del catalizador. [67]
Por ejemplo, Ting et al. introdujo una funcionalidad azido en proteínas específicas en la superficie celular usando una enzima ligasa . A continuación, la proteína marcada con azida se marca con un conjugado de ciclooctina-fluoróforo para producir una proteína marcada con fluorescencia. [68]
Referencias
- ^ Huisgen, Rolf (1963). "1.3-Dipolare Cycloadditionen Ruckschau und Ausblick". Angewandte Chemie . 75 (13): 604–637. doi : 10.1002 / ange.19630751304 .
- ↑ a b Huisgen, Rolf (noviembre de 1963). "Cinética y mecanismo de cicloadiciones 1,3-dipolares". Angewandte Chemie International Edition . 2 (11): 633–645. doi : 10.1002 / anie.196306331 .
- ^ Firestone, R (1968). "Mecanismo de cicloadiciones 1,3-dipolares". Revista de Química Orgánica . 33 (6): 2285–2290. doi : 10.1021 / jo01270a023 .
- ^ Huisgen, Rolf (1976). "Cicloadiciones 1,3-dipolares. 76. Naturaleza concertada de las cicloadiciones 1,3-dipolares y la cuestión de los intermedios dirradicales". Revista de Química Orgánica . 41 (3): 403–419. doi : 10.1021 / jo00865a001 .
- ^ Mloston, G .; Langhals, E .; Huisgen, Rolf (1986). "Primeras cicloadidas 1,2-dipolares de dos pasos: no estereoespecificidad". Mermelada. Chem. Soc. 108 (20): 6401–66402. doi : 10.1021 / ja00280a053 .
- ^ Seyyed Amir, Siadati (2015). "Un ejemplo de un mecanismo paso a paso para la cicloadición 1,3-dipolar sin catalizador entre un óxido de nitrilo y un alqueno rico en electrones". Letras de tetraedro . 56 (34): 4857–4863. doi : 10.1016 / j.tetlet.2015.06.048 .
- ^ Huisgen, Rolf (1963). "Cicloadiciones 1,3-dipolares. Pasado y futuro". Angewandte Chemie International Edition . 2 (10): 565–598. doi : 10.1002 / anie.196305651 .
- ^ Cox, A; Thomas, L; Sheridan, J (1958). "Espectros de microondas de diazometano y sus derivados deutero". Naturaleza . 181 (4614): 1000–1001. Código Bibliográfico : 1958Natur.181.1000C . doi : 10.1038 / 1811000a0 . S2CID 4245746 .
- ^ Hilberty, P; Leforestier, C (1978). "Expansión de funciones de onda de orbitales moleculares en funciones de onda de enlace de valencia. Un procedimiento simplificado". Revista de la Sociedad Química Estadounidense . 100 (7): 2012-2017. doi : 10.1021 / ja00475a007 .
- ^ McGarrity, JF; Patai, Saul (1978). Basicidad, acidez y enlaces de hidrógeno . Grupos diazonio y diazo . 1 . págs. 179–230. doi : 10.1002 / 9780470771549.ch6 . ISBN 9780470771549.
- ^ Berner, Daniel; McGarrity, John (1979). "Observación directa del ion metildiazonio en ácido fluorosulfúrico". Revista de la Sociedad Química Estadounidense . 101 (11): 3135–3136. doi : 10.1021 / ja00505a059 .
- ^ Muller, Eugen; Rundel, Wolfgans (1956). "Untersuchungen an Diazomethanen, VI. Mitteil .: Umsetzung von Diazoäthan mit Methyllithium". Chemische Berichte . 89 (4): 1065–1071. doi : 10.1002 / cber.19560890436 .
- ^ Geittner, Jochen; Huisgen, Rolph; Reissig, Hans-Ulrich (1978). "Dependencia de solventes de tasas de cicloadición de fenildiazometano y parámetros de activación". Heterociclos . 11 : 109-120. doi : 10.3987 / S (N) -1978-01-0109 .
- ^ Huisgen, Rolph; Reissig, Hans-Ulrich; Huber, Helmut; Voss, Sabine (1979). "Compuestos de α-diazocarbonilo y enaminas - una dicotomía de vías de reacción". Letras de tetraedro . 20 (32): 2987–2990. doi : 10.1016 / S0040-4039 (00) 70991-9 .
- ^ Sustmann, R (1974). "Control de la energía orbital de la reactividad de la cicloadición" . Química pura y aplicada . 40 (4): 569–593. doi : 10.1351 / pac197440040569 .
- ^ Geittner, Jochen; Huisgen, Rolf (1977). "Cinética de reacciones de cicloadición 1,3-dipolar de diazometano; una correlación con energías homo-lumo". Letras de tetraedro . 18 (10): 881–884. doi : 10.1016 / S0040-4039 (01) 92781-9 .
- ^ Huisgen, Rolf; Szeimies, Gunter; Mobius, Leander (1967). "K1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen". Chemische Berichte . 100 (8): 2494–2507. doi : 10.1002 / cber.19671000806 .
- ^ Williamson, DG; Cvetanovic, RJ (1968). "Velocidades de reacciones ozono-olefinas en soluciones de tetracloruro de carbono". Revista de la Sociedad Química Estadounidense . 90 (14): 3668–3672. doi : 10.1021 / ja01016a011 .
- ^ Bihlmaier, Werner; Geittner, Jochen; Huisgen, Rolf; ReissigP, Hans-Ulrich (1978). "La estereoespecificidad de las cicloadiciones de diazometano" . Heterociclos . 10 : 147-152. doi : 10.3987 / S-1978-01-0147 .
- ^ Huisgen, Rolf; Scheer, Wolfgang; Huber, Helmut (1967). "Conversión estereoespecífica de aziridinas isoméricas cis-trans en iluros de azometina de cadena abierta". Revista de la Sociedad Química Estadounidense . 89 (7): 1753-1755. doi : 10.1021 / ja00983a052 .
- ↑ Dahmen, Alexander; Hamberger, Helmut; Huisgen, Rolf; Markowski, Volker (1971). "Apertura del anillo conrotatorio de óxidos de cianoestilbeno a iluros de carbonilo". Revista de la Sociedad Química D: Comunicaciones químicas (19): 1192-1194. doi : 10.1039 / C29710001192 .
- ^ Padwa, Albert; Smolanoff, Joel (1971). "Fotocicloadición de arilazirenos con olefinas deficientes en electrones". Revista de la Sociedad Química Estadounidense . 93 (2): 548–550. doi : 10.1021 / ja00731a056 .
- ^ Iwashita, Takashi; Kusumi, Takenori; Kakisawa, Hiroshi (1979). "Una síntesis de dl-isoretronecanol" . Letras de química . 8 (11): 1337-1340. doi : 10.1246 / cl.1979.1337 .
- ^ Wang, Chia-Lin; Ripka, William; Confalone, Pat (1984). "Una síntesis breve y estereoespecífica de (±) -α-lycorane". Letras de tetraedro . 25 (41): 4613–4616. doi : 10.1016 / S0040-4039 (01) 91213-4 .
- ^ Kanemasa, Shuji (2002). "Estereocontrol asistido por metal de reacciones de cicloadición 1,3-dipolar". Synlett . 2002 (9): 1371-1387. doi : 10.1055 / s-2002-33506 .
- ^ Bode, Jeffrey; Carreira, Erick (2011). "Síntesis estereoselectiva de epotilonas A y B vía cicloadición de óxido de nitrilo dirigida". Revista de la Sociedad Química Estadounidense . 123 (15): 3611–3612. doi : 10.1021 / ja0155635 . PMID 11472140 .
- ^ Vsevolod V. Rostovtsev; Luke G. Green; Valery V. Fokin; K. Barry Sharpless (2002). "Un proceso de cicloadición escalonada de Huisgen: ligadura regioselectiva catalizada por cobre (I) de azidas y alquinos terminales". Angewandte Chemie International Edition . 41 (14): 2596–22599. doi : 10.1002 / 1521-3773 (20020715) 41:14 <2596 :: AID-ANIE2596> 3.0.CO; 2-4 . PMID 12203546 .
- ^ Caramella, Pierluigi; Houk, KN (1976). "Geometrías de betaínas de nitrilio. El esclarecimiento de reacciones aparentemente anómalas de 1,3-dipolos". Revista de la Sociedad Química Estadounidense . 98 (20): 6397–6399. doi : 10.1021 / ja00436a062 .
- ^ Caramella, Pierluigi; Gandour, Ruth W .; Hall, Janet A .; Deville, Cynthia G .; Houk, KN (1977). "Una derivación de las formas y energías de los orbitales moleculares de 1,3-dipolos. Optimizaciones geométricas de estas especies por MINDO / 2 y MINDO / 3". Revista de la Sociedad Química Estadounidense . 99 (2): 385–392. doi : 10.1021 / ja00444a013 .
- ^ Huisgen, Rolf (noviembre de 1963). "Cinética y mecanismo de cicloadiciones 1,3-dipolares". Angewandte Chemie International Edition . 2 (11): 633–645. doi : 10.1002 / anie.196306331 .
- ^ Padwa, Albert (1983). Química de cicloadición 1,3-dipolar . Serie de química heterocíclica general. 1 . Estados Unidos de América: Wiley-Interscience. págs. 141-145. ISBN 978-0-471-08364-1.
- ^ Koszinowski, J. (1980). tesis (tesis doctoral).
- ^ Evans, David; Ripin, David; Halstead, David; Campos, Kevin (1999). "Síntesis y asignación estereoquímica absoluta de (+) - Miyakolide". Revista de la Sociedad Química Estadounidense . 121 (29): 6816–6826. doi : 10.1021 / ja990789h .
- ^ Reacciones sintéticas de enlaces M = C y M = N: formación, reordenamiento y cicloadición 1,3-dipolar de Ylide; Hiyama, TW, J., Ed .; Elsevier, 2007; Vol. 11.
- ^ Padwa, Albert .; Hornbuckle, Susan F. (1991). "Formación de iluros a partir de la reacción de carbenos y carbenoides con pares solitarios de heteroátomos". Revisiones químicas . 91 (3): 263-309. doi : 10.1021 / cr00003a001 .
- ↑ a b Janulis, Eugene P .; Arduengo, Anthony J. (1983). "Estructura de un iluro de carbonilo estabilizado electrónicamente". Revista de la Sociedad Química Estadounidense . 105 (18): 5929–5930. doi : 10.1021 / ja00356a044 .
- ^ Prakash, GKS; Ellis, RW; Felberg, JD; Olah, GA Formaldehído 0-metiluro, [CH2 = O + -CH2 : El iluro de carbonilo original] J Am Chem Soc 1986, 108, 1341.
- ^ Sammes, PG; Street, LJ Adiciones de ciclo intramoleculares con Oxido pyrylium Ylides J. Chem. Soc., Chem. Comun. 1982, 1056.
- ^ Garst, YO; McBride, BJ; Douglass III, JG Cicloadiciones intramoleculares con 2- (ω-alquenil) -5-hidroxi-4-pironas Tetrahedron Lett. 1983, 24, 1675.
- ^ Gisch, John F .; Landgrebe, John A. (1985). "Diclorocarbeno de la pirólisis instantánea al vacío de trimetil (triclorometil) silano. Posible observación de 1,1-dicloro-3-fenilcarbonil iluro" . La Revista de Química Orgánica . 50 (12): 2050-2054. doi : 10.1021 / jo00212a009 .
- ^ Huan, Zhenwei; Landgrebe, John A .; Peterson, Kimberly (1983). "Iluros de dibromocarbonilo. Desoxigenación de aldehídos y cetonas por dibromocarbeno". La Revista de Química Orgánica . 48 (24): 4519–4523. doi : 10.1021 / jo00172a015 .
- ^ Martin, Charles W .; Lund, Paul R .; Rapp, Erich; Landgrebe, John A. (1978). "Iluros de carbonilo halogenados en las reacciones de precursores de dihalocarbeno mercurial con benzaldehídos sustituidos". La Revista de Química Orgánica . 43 (6): 1071–1076. doi : 10.1021 / jo00400a009 .
- ^ Hodgson, DM; Bruckl, T .; Glen, R .; Labande, AH; Selden, DA; Dossetter, AG; Redgrave, AJ Cicloadiciones intermoleculares enantioselectivas catalíticas de iluros de carbonilo derivados de 2-diazo-3,6-dicetoéster con alqueno dipolarófilos Actas de la Academia Nacional de Ciencias de los Estados Unidos de América 2004, 101, 5450.
- ^ Padwa, Albert; Hertzog, Donald L .; Nadler, William R. (1994). "Cicloadición intramolecular de dipolos de isomunchnone a .pi.-sistemas heteroaromáticos". La Revista de Química Orgánica . 59 (23): 7072–7084. doi : 10.1021 / jo00102a037 .
- ↑ Hamaguchi, M .; Ibata, T. Nuevo tipo de sistema mesoiónico. Cicloadición 1,3-dipolar de Isomunchnon con compuestos etilénicos Chem Lett 1975, 499.
- ^ Parque, Soon-Bong; Sakata, Naoya; Nishiyama, Hisao (1996). "Complejos de ariloxicarbonilcarbeno de bis (oxazolinil) piridinerutenio como intermedios activos en ciclopropanaciones catalíticas asimétricas". Química: una revista europea . 2 (3): 303–306. doi : 10.1002 / chem.19960020311 .
- ^ Snyder, James P .; Padwa, Albert; Stengel, Thomas; Arduengo, Anthony J .; Jockisch, Alexander; Kim, Hyo-Joong (2001). "Un carbenoide estable de tetracarboxilato de dirodio: estructura cristalina, análisis de enlace y catálisis". Revista de la Sociedad Química Estadounidense . 123 (45): 11318-11319. doi : 10.1021 / ja016928o . PMID 11697986 .
- ^ a b Hodgson, DM; Pierard, FYTM; Stupple, PA Reordenamientos enantioselectivos catalíticos y cicloadiciones que involucran iluros de compuestos diazo Chem Soc Rev 2001, 30, 50.
- ^ Yoshikai, Naohiko; Nakamura, Eiichi (2003). "Estudios teóricos sobre ciclación diastereo y enantioselectiva catalizada por rodio de diazocompuestos a través de inserción intramolecular C-H Bond". Síntesis y catálisis avanzada . 345 (910): 1159-1171. doi : 10.1002 / adsc.200303092 .
- ^ Nakamura, Eiichi; Yoshikai, Naohiko; Yamanaka, Masahiro (2002). "Mecanismo de activación de enlace C-H / reacción de formación de enlace C-C entre compuesto diazo y alcano catalizado por tetracarboxilato de dirodio". Revista de la Sociedad Química Estadounidense . 124 (24): 7181–7192. doi : 10.1021 / ja017823o . PMID 12059244 .
- ^ Costantino, G .; Rovito, R .; Macchiarulo, A .; Pellicciari, R. Estructura de intermedios de metal-carbenoides derivados de la descomposición mediada por tetracarboxilato de dirodio (II) de compuestos de α-diazocarbonilo: un estudio DFT J Mol Struc-Theochem 2002, 581, 111.
- ↑ a b c d M. Hodgson, D .; H. Labande, A .; Muthusamy, S. En Organic Reactions; John Wiley & Sons, Inc .: 2004.
- ^ Suga, Hiroyuki; Ebiura, Yasutaka; Fukushima, Kazuaki; Kakehi, Akikazu; Baba, Toshihide (2005). "Efectos catalíticos eficientes de los ácidos de Lewis en las reacciones de cicloadición 1,3-dipolar de iluros de carbonilo con iminas". La Revista de Química Orgánica . 70 (26): 10782–10791. doi : 10.1021 / jo051743b . PMID 16356001 .
- ^ a b Padwa, Albert; Fryxell, Glen E .; Zhi, Lin (1990). "Reacción tándem de ciclación-cicloadición de carbenoides de rodio. Alcance y detalles mecanicistas del proceso". Revista de la Sociedad Química Estadounidense . 112 (8): 3100–3109. doi : 10.1021 / ja00164a034 .
- ^ Houk, KN; Sims, Joyner .; Duke, RE; Strozier, RW; George, John K. (1973). "Orbitales moleculares de frontera de 1,3 dipolos y dipolarófilos". Revista de la Sociedad Química Estadounidense . 95 (22): 7287–7301. doi : 10.1021 / ja00803a017 .
- ^ Houk, KN; Rondan, Nelson G .; Santiago, Cielo; Gallo, Catherine J .; Gandour, Ruth Wells; Griffin, Gary W. (1980). "Estudios teóricos de las estructuras y reacciones de iluros de carbonilo sustituidos". Revista de la Sociedad Química Estadounidense . 102 (5): 1504-1512. doi : 10.1021 / ja00525a006 .
- ^ Padwa, Albert; Weingarten, M. David (1996). "Procesos en cascada de metalo carbenoides". Revisiones químicas . 96 (1): 223–270. doi : 10.1021 / cr950022h . PMID 11848752 .
- ^ Padwa, Albert; Austin, David J. (1996). "Selectividad inducida por ligando en las reacciones catalizadas por rodio (II) de compuestos α-diazocarbonilo †". La Revista de Química Orgánica . 61 : 63–72. doi : 10.1021 / jo951576n .
- ^ Suga, H .; Kakehi, A .; Ito, S .; Inoue, K .; Ishida, H .; Ibata, T. Stereocontrol en una reacción de cicloadición 1,3-dipolar catalizada por triflato de iterbio de iluro de carbonilo con maleimidas N-sustituidas y fumarato de dimetilo B Chem Soc Jpn 2001, 74, 1115.
- ^ Geng, Zhe; Chen, Bin; Chiu, Pauline (2006). "Síntesis total de ácido pseudolarico A". Angewandte Chemie International Edition . 45 (37): 6197–6201. doi : 10.1002 / anie.200602056 . PMID 16906616 .
- ^ Suga, Hiroyuki; Inoue, Kei; Inoue, Shuichi; Kakehi, Akikazu; Shiro, Motoo (2005). "Complejos de metales de tierras raras 2,6-bis (oxazolinil) piridina quiral como catalizadores para reacciones de cicloadición 1,3-dipolar altamente enantioselectivas de 2-benzopirilio-4-olatos". La Revista de Química Orgánica . 70 (1): 47–56. doi : 10.1021 / jo049007f . PMID 15624905 .
- ^ Onishi, Tomoyuki; Sebahar, Paul; Williams, Robert (2003). "Síntesis total asimétrica y concisa de espirotriprostatina A". Letras orgánicas . 5 (17): 3135–3137. doi : 10.1021 / ol0351910 . PMID 12917000 .
- ^ Tornoe, Christian; Christensen, Caspar; Meldal, Morten (2002). "Peptidotriazoles en fase sólida: [1,2,3] -triazoles por cicloadiciones 1,3-dipolares de alquinos terminales a azidas catalizadas por cobre (I) regioespecíficas". Revista de Química Orgánica . 67 (9): 3057–3064. doi : 10.1021 / jo011148j . PMID 11975567 .
- ^ Rostovtsev, Vsevolod; Green, Luke; Fokin, Valery; Sharpless, Barry K. (2002). "Un proceso de cicloadición escalonada de Huisgen: ligadura regioselectiva catalizada por cobre (I) de azidas y alquinos terminales". Angewandte Chemie International Edition . 41 (14): 2596-2599. doi : 10.1002 / 1521-3773 (20020715) 41:14 <2596 :: AID-ANIE2596> 3.0.CO; 2-4 . PMID 12203546 .
- ^ Besanceney-Webler, Christen; Jiang, Hao; Zheng, Tianqing; Feng, Lei; Soriano del Amo, David; Wang, Wei; Klivansky, Liana M .; Marlow, Florence L .; Liu, Yi; Wu, Peng (2011). "Aumento de la eficacia de las reacciones de clic bioortogonal para la bioconjugación: un estudio comparativo" . Angewandte Chemie International Edition . 50 (35): 8051–8056. doi : 10.1002 / anie.201101817 . PMC 3465470 . PMID 21761519 .
- ^ Breidenbach, Mark; Gallagher, Jennifer; Rey David; Inteligente, Brian; Wu, Peng; Bertozzi, Carolyn (2010). "Etiquetado metabólico dirigido de levaduras N-glicanos con azúcares no naturales" . Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 107 (9): 3988–3993. Código Bibliográfico : 2010PNAS..107.3988B . doi : 10.1073 / pnas.0911247107 . PMC 2840165 . PMID 20142501 .
- ↑ Agard, Nicholas; Prescher, Jennifer; Bertozzi, Carolyn (2004). "Una cicloadición azida-alquino [3 + 2] promovida por cepas para la modificación covalente de biomoléculas en sistemas vivos". Revista de la Sociedad Química Estadounidense . 126 (46): 15046–15047. doi : 10.1021 / ja044996f . PMID 15547999 .
- ↑ Fernandez-Suarez, Marta; Baruah, Hemanta; Martínez-Hernández, Laura; Xie, Kathleen; Baskin, Jeremy; Bertozzi, Carolyn; Ting, Alice (2007). "Redirigir la ligasa de ácido lipoico para el etiquetado de proteínas de la superficie celular con sondas de moléculas pequeñas" . Biotecnología de la naturaleza . 25 (12): 1483–1487. doi : 10.1038 / nbt1355 . PMC 2654346 . PMID 18059260 .