| CFTR | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
  | |||||||||||||||||||||||||
| |||||||||||||||||||||||||
| Identificadores | |||||||||||||||||||||||||
| Alias | CFTR , ABC35, ABCC7, CF, CFTR / MRP, MRP7, TNR-dJ760C5.1, regulador de conductancia transmembrana de fibrosis quística, regulador de conductancia transmembrana CF | ||||||||||||||||||||||||
| Identificaciones externas | OMIM : 602421 MGI : 88388 HomoloGene : 55465 GeneCards : CFTR | ||||||||||||||||||||||||
| Número CE | 5.6.1.6 | ||||||||||||||||||||||||
| |||||||||||||||||||||||||
| |||||||||||||||||||||||||
| |||||||||||||||||||||||||
| Ortólogos | |||||||||||||||||||||||||
| Especies | Humano | Ratón | |||||||||||||||||||||||
| Entrez |
|
| |||||||||||||||||||||||
| Ensembl |
|
| |||||||||||||||||||||||
| UniProt |
|
| |||||||||||||||||||||||
| RefSeq (ARNm) |
|
| |||||||||||||||||||||||
| RefSeq (proteína) |
|
| |||||||||||||||||||||||
| Ubicación (UCSC) | Crónicas 7: 117,29 - 117,72 Mb | Crónicas 6: 18,17 - 18,32 Mb | |||||||||||||||||||||||
| Búsqueda en PubMed | [3] | [4] | |||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||
| |||||||||||||||||||||||||




El regulador de la conductancia transmembrana de la fibrosis quística ( CFTR ) es una proteína de membrana y un canal de cloruro en los vertebrados que está codificado por el gen CFTR . [5] [6]
El gen CFTR codifica una proteína de canal iónico de clase transportadora ABC que conduce iones cloruro [7] a través de las membranas de las células epiteliales . Las mutaciones del gen CFTR que afectan la función del canal de iones cloruro conducen a una desregulación del transporte de líquido epitelial en el pulmón, páncreas y otros órganos, lo que resulta en fibrosis quística . Las complicaciones incluyen moco espesado en los pulmones con frecuentes infecciones respiratorias e insuficiencia pancreática que da lugar a desnutrición y diabetes. Estas condiciones conducen a una discapacidad crónica y una reducción de la esperanza de vida. En los pacientes masculinos, la obstrucción progresiva y la destrucción de los conductos deferentes en desarrollo (cordón espermático) y el epidídimo parecen ser el resultado de secreciones intraluminales anormales, [8] que causan ausencia congénita de los conductos deferentes e infertilidad masculina.
Gene

El gen que codifica la proteína CFTR humana se encuentra en el cromosoma 7 , en el brazo largo en la posición q31.2. [6] desde el par de bases 116.907.253 al par de bases 117.095.955. Los ortólogos CFTR [9] ocurren en los vertebrados con mandíbula . [10]
El gen CFTR se ha utilizado en animales como marcador filogenético de ADN nuclear . [9] Se han utilizado grandes secuencias genómicas de este gen para explorar la filogenia de los principales grupos de mamíferos , [11] y se confirmó la agrupación de órdenes placentarios en cuatro clados principales: Xenarthra , Afrotheria , Laurasiatheria y Euarchonta más Glires .
Mutaciones
Se han descrito casi 1000 mutaciones causantes de fibrosis quística . [12] La mutación más común, DeltaF508 (ΔF508) resulta de una deleción (Δ) de tres nucleótidos que resulta en una pérdida del aminoácido fenilalanina (F) en la posición 508 de la proteína. [13] Como resultado, la proteína no se pliega normalmente y se degrada más rápidamente. La gran mayoría de mutaciones son poco frecuentes. La distribución y frecuencia de las mutaciones varía entre las diferentes poblaciones, lo que tiene implicaciones para la detección y el asesoramiento genéticos.
El descubrimiento de fármacos con fines terapéuticos para abordar la FQ en todos los pacientes es complicado debido a la gran cantidad de mutaciones que causan enfermedades. Idealmente, se requiere una biblioteca de líneas celulares y ensayos basados en células correspondientes a todos los mutantes para seleccionar fármacos candidatos ampliamente activos. Pueden usarse métodos de ingeniería celular que incluyen sondas de señalización de oligonucleótidos fluorogénicos para detectar y aislar líneas celulares clonales para cada mutante. [14]
Las mutaciones consisten en reemplazos, duplicaciones, deleciones o acortamientos en el gen CFTR. Esto puede resultar en proteínas que pueden no funcionar, trabajar con menos eficacia, degradarse más rápidamente o estar presentes en cantidades inadecuadas. [15]
Se ha planteado la hipótesis de que las mutaciones en el gen CFTR pueden conferir una ventaja selectiva a los individuos heterocigotos. Las células que expresan una forma mutante de la proteína CFTR son resistentes a la invasión de la bacteria Salmonella typhi , el agente de la fiebre tifoidea , y los ratones que portan una sola copia de CFTR mutante son resistentes a la diarrea causada por la toxina del cólera. [dieciséis]
Las mutaciones más comunes que causan fibrosis quística e insuficiencia pancreática en humanos son: [17]
| Nombre de la variante de ADNc (ordenados de 5 'a 3') | Nombre de la proteína variante | Nombre de la variante heredada | rsID | # alelos en CFTR2 | Frecuencia alélica en CFTR2 | % pancreático insuficiente | Determinación final variante (julio de 2020) |
| c.1521_1523delCTT | p.Phe508del | F508del | rs113993960 | 99061 | 0,69744 | 98% | Causante de fibrosis quística |
| c.1624G> T | p.Gly542X | G542X | rs113993959 | 3610 | 0.02542 | 98% | Causante de fibrosis quística |
| c.1652G> A | p.Gly551Asp | G551D | rs75527207 | 2986 | 0.02102 | 96% | Causante de fibrosis quística |
| c.3909C> G | p.Asn1303Lys | N1303K | rs80034486 | 2246 | 0.01581 | 98% | Causante de fibrosis quística |
| c.350G> A | p.Arg117His | R117H | rs78655421 | 1854 | 0.01305 | 23% | Consecuencia clínica variable |
| c.3846G> A | p.Trp1282X | W1282X | rs77010898 | 1726 | 0.01215 | 99% | Causante de fibrosis quística |
| c.489 + 1G> T | Sin nombre de proteína | 621 + 1G-> T | rs78756941 | 1323 | 0,00931 | 99% | Causante de fibrosis quística |
| c.1657C> T | p.Arg553X | R553X | rs74597325 | 1323 | 0,00931 | 97% | Causante de fibrosis quística |
| c.1585-1G> A | Sin nombre de proteína | 1717-1G-> A | rs76713772 | 1216 | 0,00856 | 97% | Causante de fibrosis quística |
| c.3718-2477C> T | Sin nombre de proteína | 3849 + 10 kbC-> T | rs75039782 | 1158 | 0,00815 | 33% | Causante de fibrosis quística |
| c.2657 + 5G> A | Sin nombre de proteína | 2789 + 5G-> A | rs80224560 | 1027 | 0,00723 | 43% | Causante de fibrosis quística |
| c.1519_1521delATC | p.Ile507del | I507del | rs121908745 | 651 | 0,00458 | 98% | Causante de fibrosis quística |
| c.3484C> T | p.Arg1162X | R1162X | rs74767530 | 651 | 0,00458 | 97% | Causante de fibrosis quística |
| c.254G> A | p.Gly85Glu | G85E | rs75961395 | 616 | 0,00434 | 85% | Causante de fibrosis quística |
| c.3454G> C | p.Asp1152His | D1152H | rs75541969 | 571 | 0,00402 | 24% | Consecuencia clínica variable |
| c.2051_2052delAAinsG | p.Lys684SerfsX38 | 2183AA-> G | rs121908799 | 542 | 0,00382 | 96% | Causante de fibrosis quística |
| c.3528delC | p.Lys1177SerfsX15 | 3659delC | rs121908747 | 539 | 0,00379 | 99% | Causante de fibrosis quística |
| c.1040G> C | p.Arg347Pro | R347P | rs77932196 | 533 | 0,00375 | 68% | Causante de fibrosis quística |
| c.1210-12T [5] | Sin nombre de proteína | 5T | rs1805177 | 516 | 0,00363 | 28% | Consecuencia clínica variable |
| c.2988 + 1G> A | Sin nombre de proteína | 3120 + 1G-> A | rs75096551 | 501 | 0,00353 | 98% | Causante de fibrosis quística |
| c.1364C> A | p.Ala455Glu | A455E | rs74551128 | 500 | 0,00352 | 34% | Causante de fibrosis quística |
| c.3140-26A> G | Sin nombre de proteína | 3272-26A-> G | rs76151804 | 470 | 0,00331 | 29% | Causante de fibrosis quística |
| c.1000C> T | p.Arg334Trp | R334W | rs121909011 | 429 | 0,00302 | 40% | Causante de fibrosis quística |
| hacia 1766 + 1G> A | Sin nombre de proteína | 1898 + 1G-> A | rs121908748 | 421 | 0,00296 | 99% | Causante de fibrosis quística |
| c.54-5940_273 + 10250del21kb | p.Ser18ArgfsX16 | CFTRdele2,3 | extraviado | 417 | 0,00294 | 100% | Causante de fibrosis quística |
| c.1679G> C | p.Arg560Thr | R560T | rs80055610 | 343 | 0,00241 | 98% | Causante de fibrosis quística |
| c.617T> G | p.Leu206Trp | L206W | rs121908752 | 333 | 0,00234 | 20% | Causante de fibrosis quística |
| c.2052dupA | p.Gln685ThrfsX4 | 2184insA | rs121908786 | 329 | 0,00232 | 85% | Causante de fibrosis quística |
| c.262_263delTT | p.Leu88IlefsX22 | 394delTT | rs121908769 | 307 | 0,00216 | 97% | Causante de fibrosis quística |
| c.178G> T | p.Glu60X | E60X | rs77284892 | 296 | 0,00208 | 99% | Causante de fibrosis quística |
| c.1477C> T | p.Gln493X | Q493X | rs77101217 | 292 | 0,00206 | 98% | Causante de fibrosis quística |
| c.579 + 1G> T | Sin nombre de proteína | 711 + 1G-> T | rs77188391 | 274 | 0,00193 | 98% | Causante de fibrosis quística |
| c.2052delA | p.Lys684AsnfsX38 | 2184delA | rs121908746 | 255 | 0,00180 | 98% | Causante de fibrosis quística |
| c.200C> T | p.Pro67Leu | P67L | rs368505753 | 239 | 0,00168 | 34% | Causante de fibrosis quística |
| c.3302T> A | p.Met1101Lys | M1101K | rs36210737 | 238 | 0,00168 | 69% | Causante de fibrosis quística |
| c.1408A> G | p.Met470Val | M470V | rs213950 | 235 | 0,00165 | 46% | No causa fibrosis quística |
| c.3276C> A o c.3276C> G | p.Tyr1092X | Y1092X | rs121908761 | 225 | 0,00158 | 98% | Causante de fibrosis quística |
| c.3196C> T | p.Arg1066Cys | R1066C | rs78194216 | 220 | 0,00155 | 98% | Causante de fibrosis quística |
| c.1021_1022dupTC | p.Phe342HisfsX28 | 1154insTC | rs387906360 | 214 | 0,00151 | 99% | Causante de fibrosis quística |
| c.3773dupT | p.Leu1258PhefsX7 | 3905insT | rs121908789 | 210 | 0,00148 | 97% | Causante de fibrosis quística |
| c.1646G> A | p.Ser549Asn | S549N | rs121908755 | 203 | 0,00143 | 84% | Causante de fibrosis quística |
| c.1040G> A | p.Arg347His | R347H | rs77932196 | 199 | 0,00140 | 24% | Causante de fibrosis quística |
| c.948delT | p.Phe316LeufsX12 | 1078delT | rs121908744 | 184 | 0,00130 | 99% | Causante de fibrosis quística |
| c.1210-33_1210-6GT [12] T [4] | Sin nombre de proteína | 5T; TG12 | extraviado | 182 | 0,00128 | 14% | Consecuencia clínica variable |
| c.3472C> T | p.Arg1158X | R1158X | rs79850223 | 179 | 0,00126 | 99% | Causante de fibrosis quística |
| c.2834C> T | p.Ser945Leu | S945L | rs397508442 | 167 | 0,00118 | 40% | Causante de fibrosis quística |
| c.1558G> T | p.Val520Phe | V520F | rs77646904 | 156 | 0,00110 | 98% | Causante de fibrosis quística |
| c.443T> C | p.Ile148Thr | I148T | rs35516286 | 148 | 0,00104 | 88% | No causa fibrosis quística |
| c.349C> T | p.Arg117Cys | R117C | rs77834169 | 146 | 0,00103 | 24% | Causante de fibrosis quística |
DeltaF508
DeltaF508 ( ΔF508 ), nombre completo CFTRΔF508 o F508del-CFTR ( rs113993960 ), es una mutación específica dentro del gen CFTR que implica una deleción de tres nucleótidos que abarcan las posiciones 507 y 508 del gen CFTR en el cromosoma 7, que finalmente resulta en la pérdida de un solo codón para el aminoácido fenilalanina (F). Una persona con la mutación CFTRΔF508 producirá una proteína CFTR anormal que carece de este residuo de fenilalanina y que no puede plegarse correctamente. Esta proteína no escapa del retículo endoplásmico.para su posterior procesamiento. Tener dos copias de esta mutación (una heredada de cada padre) es, con mucho, la causa más común de fibrosis quística (FQ), responsable de casi dos tercios de las mutaciones en todo el mundo. [18]
Efectos
La proteína CFTR se expresa en gran medida en las células del páncreas, el epitelio intestinal y respiratorio y todas las glándulas exocrinas. Cuando se pliega correctamente, se transporta a la membrana celular, donde se convierte en una proteína transmembrana responsable de abrir los canales que liberan iones de cloruro de las células; también inhibe simultáneamente la captación de iones de sodio por otra proteína de canal. Ambas funciones ayudan a mantener un gradiente de iones que hace que la ósmosis extraiga agua de las células. [19] La mutación ΔF508 conduce al plegamiento incorrecto de CFTR y su eventual degradación en el RE.. En organismos con dos complementos de la mutación, la proteína está completamente ausente de la membrana celular y estas funciones críticas de transporte de iones no se llevan a cabo. [20]
Tener un par de genes homocigotos con la mutación ΔF508 evita que la proteína CFTR asuma su posición normal en la membrana celular. Esto provoca una mayor retención de agua en las células, la correspondiente deshidratación del espacio extracelular y una cascada de efectos asociada en varias partes del cuerpo. Estos efectos incluyen: membranas mucosas más gruesas en el epitelio de los órganos afectados; obstrucción de las vías respiratorias estrechas como resultado de una mucosa más espesa y la inhibición del libre movimiento de las mucocilias; ausencia congénita de los conductos deferentesdebido al aumento del grosor del moco durante el desarrollo fetal; insuficiencia pancreática debido al bloqueo del conducto pancreático con moco; y un mayor riesgo de infección respiratoria debido a la acumulación de moco espeso y rico en nutrientes donde prosperan las bacterias. Estos son los síntomas de la fibrosis quística , un trastorno genético; sin embargo, ΔF508 no es la única mutación que causa este trastorno.
Ser un portador heterocigoto (que tiene una sola copia de ΔF508) da como resultado una menor pérdida de agua durante la diarrea porque las proteínas CFTR que funcionan mal o ausentes no pueden mantener gradientes iónicos estables a través de las membranas celulares. Por lo general, hay una acumulación de iones Cl - y Na + dentro de las células afectadas, lo que crea un efecto hipotónico.solución fuera de las células y haciendo que el agua se difunda dentro de las células por ósmosis. Varios estudios indican que los portadores heterocigotos tienen un mayor riesgo de sufrir diversos síntomas. Por ejemplo, se ha demostrado que la heterocigosidad para la fibrosis quística se asocia con una mayor reactividad de las vías respiratorias y los heterocigotos pueden tener riesgo de mala función pulmonar. Se ha demostrado que los heterocigotos con sibilancias tienen un mayor riesgo de mala función pulmonar o desarrollo y progresión de enfermedad pulmonar obstructiva crónica . Un gen de la fibrosis quística es suficiente para producir anomalías pulmonares leves incluso en ausencia de infección. [21]
Mecanismo
El gen CFTR se encuentra en el brazo largo del cromosoma 7, en la posición q31.2, y finalmente codifica una secuencia de 1480 aminoácidos. Normalmente, los tres pares de bases de ADN ATC (emparejados con TAG en la hebra opuesta) en la posición 507 del gen forman la plantilla para el codón de ARNm AUC para isoleucina , mientras que los tres pares de bases de ADN TTT (emparejados con AAA) en la posición 508 adyacente forman la plantilla para el codón UUU de fenilalanina . [22] La mutación ΔF508 es una deleción del par CG de la posición 507 junto con los dos primeros pares de TA de la posición 508, dejando la secuencia de ADN ATT (emparejada con TAA) en la posición 507, que se transcribeen el codón de ARNm AUU. Dado que AUU también codifica isoleucina, el aminoácido de la posición 507 no cambia y el efecto neto de la mutación es equivalente a una deleción ("Δ") de la secuencia que da como resultado el codón de fenilalanina en la posición 508. [23]
Prevalencia
ΔF508 está presente en al menos una copia del cromosoma 7 en aproximadamente uno de cada 30 caucásicos . La presencia de la mutación en ambas copias provoca la enfermedad autosómica recesiva fibrosis quística. Los científicos han estimado que la mutación original ocurrió hace más de 52.000 años en el norte de Europa . La edad del alelo joven puede ser consecuencia de una selección pasada. Una hipótesis de por qué la selección natural ha mantenido la mutación, que de otro modo sería perjudicial, es que una sola copia puede presentar un efecto positivo al reducir la pérdida de agua durante el cólera , aunque la introducción del patógeno Vibrio cholerae en Europa no se produjo hasta finales del siglo XVIII. [24]Otra teoría postula que los portadores de FQ (heterocigotos para ΔF508) son más resistentes a la fiebre tifoidea , ya que se ha demostrado que CFTR actúa como un receptor para que la bacteria Salmonella typhi ingrese a las células epiteliales intestinales. [25]
Los heterocigotos ΔF508 de fibrosis quística pueden estar sobrerrepresentados entre las personas con asma y pueden tener una función pulmonar más deficiente que los no portadores. [26] [27] Los portadores de una sola mutación de la FQ tienen una prevalencia más alta de rinosinusitis crónica que la población general. [28] Aproximadamente el 50% de los casos de fibrosis quística en Europa se deben a mutaciones homocigóticas de ΔF508 (esto varía mucho según la región), [29] mientras que la frecuencia alélica de ΔF508 es de aproximadamente el 70%. [30]Los casos restantes son causados por más de 1,500 otras mutaciones, incluidas R117H, 1717-1G> A y 2789 + 56G> A. Estas mutaciones, cuando se combinan entre sí o incluso con una sola copia de ΔF508, pueden causar síntomas de FQ. El genotipo no está fuertemente correlacionado con la gravedad de la FQ, aunque se han relacionado síntomas específicos con ciertas mutaciones.
Estructura

El gen CFTR tiene aproximadamente 189 kb de longitud, con 27 exones y 26 intrones . [31] CFTR es una glicoproteína con 1480 aminoácidos . La proteína consta de cinco dominios. Hay dos dominios transmembrana, cada uno con seis tramos de hélices alfa . Cada uno de ellos está conectado a un dominio de unión a nucleótidos (NBD) en el citoplasma. El primer NBD está conectado al segundo dominio transmembrana mediante un dominio "R" regulador que es una característica única de CFTR, no presente en otros transportadores ABC . El canal iónico solo se abre cuando su dominio R ha sido fosforilado por PKA y ATPestá vinculado a los NBD. [32] El carboxilo terminal de la proteína está anclado al citoesqueleto por un dominio de interacción PDZ . [33] La estructura mostrada (PDB # 1XMI) muestra un ensamblaje homopentamérico de NBD1 mutado, el primer dominio de unión a nucleótidos (NBD1) del transportador.
Ubicación y función
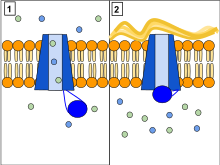
Funciones CFTR como una fosforilación y ATP - gated anión canal , aumentando la conductancia para ciertos aniones (por ejemplo, Cl - ) fluya hacia abajo de su gradiente electroquímico . Los cambios conformacionales impulsados por ATP en CFTR abren y cierran una puerta para permitir el flujo transmembrana de aniones por su gradiente electroquímico . [34] Esto en contraste con otras proteínas ABC , en las que los cambios conformacionales impulsados por ATP alimentan el transporte de sustrato cuesta arriba a través de las membranas celulares. Esencialmente, CFTR es un canal de iones que evolucionó como un 'roto'Transportador ABC que gotea cuando está en conformación abierta .
Los CFTR tienen dos dominios transmembrana, cada uno vinculado a un dominio de unión a nucleótidos. CFTR también contiene otro dominio llamado dominio regulatorio. Otros miembros de la superfamilia de transportadores ABC están involucrados en la absorción de nutrientes en procariotas o en la exportación de una variedad de sustratos en eucariotas. Los transportadores ABC han evolucionado para transducir la energía libre de la hidrólisis de ATP al movimiento ascendente de sustratos a través de la membrana celular. Tienen dos conformaciones principales, una en la que el sitio de unión de la carga está orientado hacia el citosol o hacia adentro (libre de ATP) y otra en la que está orientado hacia afuera (ligado a ATP). El ATP se une a cada dominio de unión de nucleótidos, lo que da como resultado la posterior dimerización de NBD, lo que conduce al reordenamiento de las hélices transmembrana.Esto cambia la accesibilidad del sitio de encuadernación de la carga de una posición orientada hacia adentro a una que mira hacia afuera. La unión de ATP, y la hidrólisis que sigue, impulsa la exposición alternativa del sitio de unión de la carga, asegurando un transporte unidireccional de la carga contra ungradiente electroquímico . En CFTR, alternar entre una conformación que mira hacia adentro y una que mira hacia afuera da como resultado la compuerta del canal. En particular, la dimerización de NBD (favorecida por la unión de ATP) se acopla a la transición a una conformación orientada hacia afuera en la que se forma una ruta transmembrana abierta para los aniones. La hidrólisis posterior (en el sitio canónico activo, sitio 2, incluidos los motivos de Walker de NBD2) desestabiliza el dímero NBD y favorece el retorno a la conformación orientada hacia adentro, en la que se cierra la vía de permeación de aniones. [34]
El CFTR se encuentra en las células epiteliales de muchos órganos, incluidos el pulmón , el hígado , el páncreas , el tracto digestivo y el tracto reproductivo femenino [35] y masculino . [36] [37]
En las vías respiratorias del pulmón, el CFTR se expresa en mayor medida por células especializadas poco comunes llamadas ionocitos pulmonares . [38] [39] [40] En la piel, el CFTR se expresa fuertemente en las glándulas sudoríparas sebáceas y ecrinas . [41] En las glándulas ecrinas, CFTR se encuentra en la membrana apical de las células epiteliales que forman el conducto de estas glándulas sudoríparas. [41]
Normalmente, la proteína permite el movimiento de iones cloruro y tiocianato [42] (con carga negativa) desde una célula epitelial hacia el líquido y el moco de la superficie de las vías respiratorias . Los iones de sodio cargados positivamente siguen pasivamente, aumentando la concentración total de electrolitos en el moco, lo que resulta en el movimiento del agua fuera de la célula a través de la ósmosis .
En las células epiteliales con cilios móviles que recubren el bronquio y el oviducto, el CFTR se encuentra en la membrana celular apical pero no en los cilios. [35] Por el contrario, ENaC (canal de sodio epitelial) se encuentra a lo largo de toda la longitud de los cilios. [35]
En las glándulas sudoríparas , el CFTR defectuoso da como resultado una reducción del transporte de cloruro de sodio y tiocianato de sodio [43] en el conducto de reabsorción y, por lo tanto, un sudor más salado. Esta es la base de una prueba de sudor clínicamente importante para la fibrosis quística que a menudo se usa para el diagnóstico con el cribado genético. [44]
Interacciones
Se ha demostrado que el regulador de la conductancia transmembrana de la fibrosis quística interactúa con:
- DNAJC5 , [45]
- GOPC , [46] [47]
- PDZK1 , [47] [48]
- PRKCE , [49]
- SLC4A8 , [50]
- SNAP23 , [51]
- SLC9A3R1 , [33] [50] [52] [53] [54] [55]
- SLC9A3R2 , [56] y
- STX1A , [51] [57]
Es inhibido por el crofelemer, un fármaco antidiarreico .
Condiciones relacionadas
- Ausencia congénita bilateral de conductos deferentes : los hombres con ausencia congénita bilateral de conductos deferentes suelen tener una mutación leve (un cambio que permite la función parcial del gen) en una copia del gen CFTR y una mutación que causa fibrosis quística en la otra. copia de CFTR.
- Fibrosis quística : se han encontrado más de 1.800 mutaciones en el gen CFTR [58], pero la mayoría de ellas no se han asociado con fibrosis quística. [59] La mayoría de estas mutaciones sustituyen un aminoácido (un componente básico de las proteínas) por otro aminoácido en la proteína CFTR o eliminan una pequeña cantidad de ADN.en el gen CFTR. La mutación más común, denominada ΔF508, es una deleción (Δ) de un aminoácido (fenilalanina) en la posición 508 de la proteína CFTR. Esta proteína alterada nunca llega a la membrana celular porque se degrada poco después de su fabricación. Todas las mutaciones que causan enfermedades en el gen CFTR impiden que el canal funcione correctamente, lo que provoca un bloqueo del movimiento de la sal y el agua dentro y fuera de las células. Como resultado de este bloqueo, las células que recubren los conductos de los pulmones, el páncreas y otros órganos producen una mucosidad anormalmente espesa y pegajosa. Este moco obstruye las vías respiratorias y las glándulas, provocando los signos y síntomas característicos de la fibrosis quística. Además, los cilios solo pueden eliminar la mucosidad fina ; el moco espeso no puede, por lo que atrapa las bacterias que dan lugar a infecciones crónicas.
- Cólera : la ribosilación de ADP causada por la toxina del cólera da como resultado un aumento de la producción de AMP cíclico que a su vez abre el canal CFTR que conduce a la secreción excesiva de Cl - . El Na + y el H 2 O siguen al Cl - hacia el intestino delgado, lo que resulta en deshidratación y pérdida de electrolitos. [60]
Objetivo farmacológico
CFTR ha sido un objetivo farmacológico en los esfuerzos por encontrar tratamientos para afecciones relacionadas. Ivacaftor (nombre comercial Kalydeco , desarrollado como VX-770 ) es un medicamento aprobado por la FDA en 2012 para personas con fibrosis quística que tienen mutaciones específicas de CFTR. [61] [62] Ivacaftor fue desarrollado por Vertex Pharmaceuticals junto con la Cystic Fibrosis Foundation y es el primer fármaco que trata la causa subyacente en lugar de los síntomas de la enfermedad. [63] Llamada "la nueva droga más importante de 2012", [64] y "una droga maravillosa" [65]es uno de los medicamentos más caros, con un costo de más de US $ 300.000 por año, lo que ha llevado a críticas a Vertex por su alto costo.
Referencias
- ^ a b c GRCh38: Ensembl release 89: ENSG00000001626 - Ensembl , mayo de 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000041301 - Ensembl , mayo de 2017
- ^ "Referencia humana de PubMed:" . Centro Nacional de Información Biotecnológica, Biblioteca Nacional de Medicina de EE. UU .
- ^ "Referencia de PubMed del ratón:" . Centro Nacional de Información Biotecnológica, Biblioteca Nacional de Medicina de EE. UU .
- ^ Gadsby DC, Vergani P, Csanády L (marzo de 2006). "La proteína ABC convertida en canal de cloruro cuyo fallo provoca fibrosis quística" . Naturaleza . 440 (7083): 477–83. Código Bib : 2006Natur.440..477G . doi : 10.1038 / nature04712 . PMC 2720541 . PMID 16554808 .
- ^ a b Collins F , Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, et al. (Septiembre de 1989). "Identificación del gen de la fibrosis quística: cromosoma caminando y saltando". Ciencia . 245 (4922): 1059–65. Código Bibliográfico : 1989Sci ... 245.1059R . doi : 10.1126 / science.2772657 . PMID 2772657 .
- ^ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. (Septiembre de 1989). "Identificación del gen de la fibrosis quística: clonación y caracterización de ADN complementario". Ciencia . 245 (4922): 1066–73. Código Bibliográfico : 1989Sci ... 245.1066R . doi : 10.1126 / science.2475911 . PMID 2475911 .
- ^ Marcorelles P, Gillet D, Friocourt G, Ledé F, Samaison L, Huguen G, Ferec C (marzo de 2012). "Expresión de la proteína reguladora de conductancia transmembrana de fibrosis quística en el sistema de conductos excretores masculinos durante el desarrollo". Patología humana . 43 (3): 390–7. doi : 10.1016 / j.humpath.2011.04.031 . PMID 21840567 .
- ^ a b "Marcador filogenético OrthoMaM: secuencia de codificación CFTR" . Archivado desde el original el 2 de marzo de 2016 . Consultado el 12 de marzo de 2010 .
- ^ Davies R, Conroy SJ, Davies WL, Potter IC, Trezise AE (19-23 de junio de 2005). "Evolución y regulación del gen de la fibrosis quística" (artículo de conferencia) . Conferencia de Biología Molecular y Evolución (MBE05) . Consultado el 28 de julio de 2014 .
- ^ Prasad AB, Allard MW, Green ED (septiembre de 2008). "Confirmación de la filogenia de los mamíferos mediante el uso de grandes conjuntos de datos de secuencia comparativa" . Biología Molecular y Evolución . 25 (9): 1795–808. doi : 10.1093 / molbev / msn104 . PMC 2515873 . PMID 18453548 .
- ^ "La traducción clínica y funcional de CFTR (CFTR2): historial de lista de variantes de CFTR2" . US CF Foundation, Johns Hopkins University, Cystic Fibrosis Centre en el Hospital for Sick Children de Toronto . Consultado el 2 de agosto de 2017 .[ enlace muerto permanente ]
- ^ Guimbellot J, Sharma J, Rowe SM (noviembre de 2017). "Hacia una terapia inclusiva con moduladores CFTR: avances y desafíos" . Neumología pediátrica . 52 (S48): S4 – S14. doi : 10.1002 / ppul.23773 . PMC 6208153 . PMID 28881097 .
- ^ Shekdar K, Langer J, Venkatachalan S, Schmid L, Anobile J, Shah P, et al. (Marzo de 2021). "Método de ingeniería celular utilizando sondas de señalización de oligonucleótidos fluorogénicos y citometría de flujo" . Cartas de biotecnología . doi : 10.1007 / s10529-021-03101-5 . PMC 7937778 . PMID 33683511 .
- ^ Rowe SM, Miller S, Sorscher EJ (mayo de 2005). "Fibrosis quística". La Revista de Medicina de Nueva Inglaterra . 352 (19): 1992–2001. doi : 10.1056 / NEJMra043184 . PMID 15888700 .
- ^ Kavic SM, Frehm EJ, Segal AS (1999). "Estudios de caso en el cólera: lecciones de historia médica y ciencia" . La Revista de Biología y Medicina de Yale . 72 (6): 393–408. PMC 2579035 . PMID 11138935 .
- ^ "CFTR2" . Consultado el 8 de julio de 2021 .
- ^ Bobadilla JL, Macek M, Fine JP, Farrell PM (junio de 2002). "Fibrosis quística: un análisis mundial de mutaciones CFTR - correlación con los datos de incidencia y aplicación a la detección" . Mutación humana . 19 (6): 575–606. doi : 10.1002 / humu.10041 . PMID 12007216 .
- ^ Verkman AS, Song Y, Thiagarajah JR (enero de 2003). "Papel del líquido de la superficie de las vías respiratorias y las glándulas submucosas en la enfermedad pulmonar por fibrosis quística". Revista estadounidense de fisiología. Fisiología celular . 284 (1): C2-15. doi : 10.1152 / ajpcell.00417.2002 . PMID 12475759 .
- ^ "Direcciones de investigación de la fibrosis quística" . Instituto Nacional de Diabetes y Enfermedades Digestivas y Renales (NIDDK).
- ^ Maurya N, Awasthi S, Dixit P (abril de 2012). "Asociación de la mutación del gen CFTR con el asma bronquial" (PDF) . The Indian Journal of Medical Research . 135 (4): 469–78. PMID 22664493 .
- ^ Informe CCDS para Consensus CDS: Informe para CCDS5773.1 (versión actual) NCBI
- ^ Bartoszewski RA, Jablonsky M, Bartoszewska S, Stevenson L, Dai Q, Kappes J, et al. (Septiembre de 2010). "Un polimorfismo de un solo nucleótido sinónimo en DeltaF508 CFTR altera la estructura secundaria del ARNm y la expresión de la proteína mutante" . La revista de química biológica . 285 (37): 28741–8. doi : 10.1074 / jbc.M110.154575 . PMC 2937902 . PMID 20628052 .
- ^ "Re: ¿Existe una conexión entre la fibrosis quística y el cólera?" .
- ^ Muelle GB, Lechada M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, et al. (Mayo de 1998). "Salmonella typhi utiliza CFTR para entrar en las células epiteliales intestinales". Naturaleza . 393 (6680): 79–82. Código Bibliográfico : 1998Natur.393 ... 79P . doi : 10.1038 / 30006 . PMID 9590693 . S2CID 5894247 .
- ^ Dahl M, Nordestgaard BG, Lange P, Tybjaerg-Hansen A (mayo de 2001). "Seguimiento de quince años de la función pulmonar en individuos heterocigotos para la deleción de fenilalanina-508 de fibrosis quística". La Revista de Alergia e Inmunología Clínica . 107 (5): 818–23. doi : 10.1067 / mai.2001.114117 . PMID 11344348 .
- ^ Dahl M, Tybjaerg-Hansen A, Lange P, Nordestgaard BG (junio de 1998). "Heterocigosidad DeltaF508 en fibrosis quística y susceptibilidad al asma". Lancet . 351 (9120): 1911–3. doi : 10.1016 / s0140-6736 (97) 11419-2 . PMID 9654257 . S2CID 22970136 .
- ^ Wang X, Kim J, McWilliams R, Cutting GR (marzo de 2005). "Mayor prevalencia de rinosinusitis crónica en portadores de una mutación de fibrosis quística" . Archivos de Otorrinolaringología – Cirugía de Cabeza y Cuello . 131 (3): 237–40. doi : 10.1001 / archotol.131.3.237 . PMID 15781764 .
- ^ Informe anual de ECFS: lo que significa para el Reino Unido Cystic Fibrosis Trust
- ^ Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T, Giménez J, et al. (Junio de 1994). "El origen de la principal mutación de la fibrosis quística (delta F508) en poblaciones europeas". Genética de la naturaleza . 7 (2): 169–75. doi : 10.1038 / ng0694-169 . PMID 7920636 . S2CID 38005421 .
- ^ Base de datos de mutaciones de fibrosis quística. "Secuencia de ADN genómico" . Archivado desde el original el 22 de agosto de 2016 . Consultado el 6 de abril de 2013 .
- ^ Sheppard DN, Welsh MJ (enero de 1999). "Estructura y función del canal de cloruro CFTR". Revisiones fisiológicas . 79 (Suplemento 1): S23-45. doi : 10.1152 / physrev.1999.79.1.S23 . PMID 9922375 .
- ^ a b DB corto, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, et al. (Julio de 1998). "Una proteína PDZ apical ancla el regulador de conductancia transmembrana de fibrosis quística al citoesqueleto" . La revista de química biológica . 273 (31): 19797–801. doi : 10.1074 / jbc.273.31.19797 . PMID 9677412 .
- ↑ a b Csanády L, Vergani P, Gadsby DC (enero de 2019). "ESTRUCTURA, PUERTA Y REGULACIÓN DEL CANAL ANION CFTR" (PDF) . Revisiones fisiológicas . 99 (1): 707–738. doi : 10.1152 / physrev.00007.2018 . PMID 30516439 .
- ↑ a b c Enuka Y, Hanukoglu I, Edelheit O, Vaknine H, Hanukoglu A (marzo de 2012). "Los canales de sodio epiteliales (ENaC) se distribuyen uniformemente en cilios móviles en el oviducto y las vías respiratorias". Histoquímica y Biología Celular . 137 (3): 339–53. doi : 10.1007 / s00418-011-0904-1 . PMID 22207244 . S2CID 15178940 .
- ^ Sharma S, Hanukoglu A, Hanukoglu I (abril de 2018). "Localización del canal de sodio epitelial (ENaC) y CFTR en el epitelio germinal de los testículos, células de Sertoli y espermatozoides". Revista de Histología Molecular . 49 (2): 195-208. doi : 10.1007 / s10735-018-9759-2 . PMID 29453757 . S2CID 3761720 .
- ^ Sharma S, Hanukoglu I (abril de 2019). "Mapeo de los sitios de localización del canal de sodio epitelial (ENaC) y CFTR en segmentos del epidídimo de mamíferos". Revista de Histología Molecular . 50 (2): 141-154. doi : 10.1007 / s10735-019-09813-3 . PMID 30659401 . S2CID 58026884 .
- ^ "Estudio de CF encuentra nuevas células llamadas ionocitos que llevan altos niveles de gen CFTR" . Noticias de fibrosis quística hoy . El 3 de agosto de 2018.
- ^ Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. (Agosto de 2018). "Una jerarquía epitelial de las vías respiratorias revisada incluye ionocitos que expresan CFTR" . Naturaleza . 560 (7718): 319–324. Código Bibcode : 2018Natur.560..319M . doi : 10.1038 / s41586-018-0393-7 . PMC 6295155 . PMID 30069044 .
- ^ Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. (Agosto de 2018). "Un atlas unicelular del epitelio de las vías respiratorias revela el ionocito pulmonar rico en CFTR" . Naturaleza . 560 (7718): 377–381. Código Bib : 2018Natur.560..377P . doi : 10.1038 / s41586-018-0394-6 . PMC 6108322 . PMID 30069046 .
- ↑ a b Hanukoglu I, Boggula VR, Vaknine H, Sharma S, Kleyman T, Hanukoglu A (junio de 2017). "Expresión del canal de sodio epitelial (ENaC) y CFTR en la epidermis humana y apéndices epidérmicos" . Histoquímica y Biología Celular . 147 (6): 733–748. doi : 10.1007 / s00418-016-1535-3 . PMID 28130590 . S2CID 8504408 .
- ^ Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Nauseef WM, et al. (Enero de 2007). "Un nuevo sistema de defensa del huésped de las vías respiratorias es defectuoso en la fibrosis quística" . Revista estadounidense de medicina respiratoria y de cuidados intensivos . 175 (2): 174–83. doi : 10.1164 / rccm.200607-1029OC . PMC 2720149 . PMID 17082494 .
- ^ Xu Y, Szép S, Lu Z (diciembre de 2009). "El papel antioxidante del tiocianato en la patogénesis de la fibrosis quística y otras enfermedades relacionadas con la inflamación" . Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 106 (48): 20515–9. Código bibliográfico : 2009PNAS..10620515X . doi : 10.1073 / pnas.0911412106 . PMC 2777967 . PMID 19918082 .
- ^ Yonei Y, Tanaka M, Ozawa Y, Miyazaki K, Tsukada N, Inada S, et al. (Abril de 1992). "Carcinoma hepatocelular primario con hipoglucemia severa: participación de factores de crecimiento similares a la insulina". El hígado . 12 (2): 90–3. doi : 10.1111 / j.1600-0676.1992.tb00563.x . PMID 1320177 .
- ^ Zhang H, Peters KW, Sun F, Marino CR, Lang J, Burgoyne RD, Frizzell RA (agosto de 2002). "La proteína de cadena de cisteína interactúa y modula la maduración del regulador de conductancia transmembrana de la fibrosis quística" . La revista de química biológica . 277 (32): 28948–58. doi : 10.1074 / jbc.M111706200 . PMID 12039948 .
- ^ Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, et al. (Febrero de 2002). "Una proteína de dominio PDZ asociada a Golgi modula la expresión de la membrana plasmática del regulador transmembrana de la fibrosis quística" . La revista de química biológica . 277 (5): 3520–9. doi : 10.1074 / jbc.M110177200 . PMID 11707463 .
- ↑ a b Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR (febrero de 2003). "El canal de cloruro de unión a PDZ ClC-3B se localiza en el Golgi y se asocia con proteínas PDZ que interactúan con el regulador de conductancia transmembrana de fibrosis quística" . La revista de química biológica . 278 (8): 6440–9. doi : 10.1074 / jbc.M211050200 . PMID 12471024 .
- ^ Wang S, Yue H, Derin RB, Guggino WB, Li M (septiembre de 2000). "La proteína accesoria facilitó la interacción CFTR-CFTR, un mecanismo molecular para potenciar la actividad del canal de cloruro" . Celular . 103 (1): 169–79. doi : 10.1016 / S0092-8674 (00) 00096-9 . PMID 11051556 . S2CID 16697781 .
- ^ Liedtke CM, Yun CH, Kyle N, Wang D (junio de 2002). "La regulación dependiente de la proteína quinasa C épsilon del regulador transmembrana de la fibrosis quística implica la unión a un receptor para la quinasa C activada (RACK1) y la unión de RACK1 al factor regulador de intercambio de Na + / H +" . La revista de química biológica . 277 (25): 22925–33. doi : 10.1074 / jbc.M201917200 . PMID 11956211 .
- ^ a b Park M, Ko SB, Choi JY, Muallem G, Thomas PJ, Pushkin A, et al. (Diciembre de 2002). "El regulador de la conductancia transmembrana de la fibrosis quística interactúa y regula la actividad del transportador de rescate de HCO3-, la isoforma 3 del cotransporte de Na + -HCO3- humano" . La revista de química biológica . 277 (52): 50503–9. doi : 10.1074 / jbc.M201862200 . PMID 12403779 .
- ^ a b Cormet-Boyaka E, Di A, Chang SY, Naren AP, Tousson A, Nelson DJ, Kirk KL (septiembre de 2002). "Los canales de cloruro de CFTR están regulados por un complejo SNAP-23 / sintaxina 1A" . Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 99 (19): 12477–82. Código Bibliográfico : 2002PNAS ... 9912477C . doi : 10.1073 / pnas.192203899 . PMC 129470 . PMID 12209004 .
- ^ Hegedüs T, Sessler T, Scott R, Thelin W, Bakos E, Váradi A, et al. (Marzo de 2003). "La fosforilación C-terminal de MRP2 modula su interacción con proteínas PDZ". Comunicaciones de investigación bioquímica y biofísica . 302 (3): 454–61. doi : 10.1016 / S0006-291X (03) 00196-7 . PMID 12615054 .
- ^ Wang S, Raab RW, Schatz PJ, Guggino WB, Li M (mayo de 1998). "El consenso de unión a péptidos del dominio NHE-RF-PDZ1 coincide con la secuencia C-terminal del regulador de conductancia transmembrana de fibrosis quística (CFTR)" . Cartas FEBS . 427 (1): 103–8. doi : 10.1016 / S0014-5793 (98) 00402-5 . PMID 9613608 . S2CID 20803242 .
- ^ Moyer BD, Duhaime M, Shaw C, Denton J, Reynolds D, Karlson KH, et al. (Septiembre de 2000). "El dominio de interacción con PDZ del regulador de la conductancia transmembrana de la fibrosis quística es necesario para la expresión funcional en la membrana plasmática apical" . La revista de química biológica . 275 (35): 27069–74. doi : 10.1074 / jbc.M004951200 . PMID 10852925 .
- ^ Hall RA, Ostedgaard LS, Premont RT, Blitzer JT, Rahman N, Welsh MJ, Lefkowitz RJ (julio de 1998). "Un motivo C-terminal que se encuentra en el receptor adrenérgico beta2, el receptor P2Y1 y el regulador de la conductancia transmembrana de la fibrosis quística determina la unión a la familia del factor regulador del intercambiador Na + / H + de proteínas PDZ" . Actas de la Academia Nacional de Ciencias de los Estados Unidos de América . 95 (15): 8496–501. Código Bibliográfico : 1998PNAS ... 95.8496H . doi : 10.1073 / pnas.95.15.8496 . PMC 21104 . PMID 9671706 .
- ^ Sun F, Hug MJ, Lewarchik CM, Yun CH, Bradbury NA, Frizzell RA (septiembre de 2000). "E3KARP media la asociación de ezrin y proteína quinasa A con el regulador de conductancia transmembrana de fibrosis quística en las células de las vías respiratorias" . La revista de química biológica . 275 (38): 29539–46. doi : 10.1074 / jbc.M004961200 . PMID 10893422 .
- ^ Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, et al. (Noviembre de 1997). "Regulación de los canales de cloruro de CFTR por sintaxina e isoformas de Munc18". Naturaleza . 390 (6657): 302–5. Código Bibliográfico : 1997Natur.390..302N . doi : 10.1038 / 36882 . PMID 9384384 . S2CID 4395005 .
- ^ Egan ME (marzo de 2016). "Genética de la fibrosis quística: implicaciones clínicas". Clínicas de Medicina Torácica . 37 (1): 9–16. doi : 10.1016 / j.ccm.2015.11.002 . PMID 26857764 .
- ^ De Boeck K, Amaral MD (agosto de 2016). "Avances en terapias para la fibrosis quística". La lanceta. Medicina respiratoria . 4 (8): 662–674. doi : 10.1016 / S2213-2600 (16) 00023-0 . PMID 27053340 .
- ^ Thiagarajah JR, Verkman AS (septiembre de 2012). "Inhibidores de CFTR para el tratamiento de enfermedades diarreicas" . Farmacología clínica y terapéutica . 92 (3): 287–90. doi : 10.1038 / clpt.2012.114 . PMC 3643514 . PMID 22850599 .
- ^ Jones AM, Helm JM (octubre de 2009). "Tratamientos emergentes en fibrosis quística". Drogas . 69 (14): 1903–10. doi : 10.2165 / 11318500-000000000-00000 . PMID 19747007 . S2CID 23344660 .
- ^ McPhail GL, Clancy JP (abril de 2013). "Ivacaftor: la primera terapia que actúa sobre la causa principal de la fibrosis quística". Drogas de hoy . 49 (4): 253–60. doi : 10.1358 / punto.2013.49.4.1940984 . PMID 23616952 .
- ^ "El estudio de fase 3 de VX-770 muestra una mejora notable en la función pulmonar entre personas con fibrosis quística con mutación G551D" . Comunicado de prensa . Fundación de Fibrosis Quística. 2011-02-23.
- ^ Herper M (27 de diciembre de 2012). "La nueva droga más importante de 2012" . Forbes .
- ^ Nocera J (18 de julio de 2014). "La droga de $ 300.000" . New York Times .
Lectura adicional
- Kulczycki LL, Kostuch M, Bellanti JA (enero de 2003). "Una perspectiva clínica de la fibrosis quística y nuevos hallazgos genéticos: relación de mutaciones CFTR con manifestaciones genotipo-fenotipo". Revista Estadounidense de Genética Médica. Parte A . 116A (3): 262–7. doi : 10.1002 / ajmg.a.10886 . PMID 12503104 . S2CID 9245855 .
- Vankeerberghen A, Cuppens H, Cassiman JJ (marzo de 2002). "El regulador de la conductancia transmembrana de la fibrosis quística: una proteína intrigante con funciones pleiotrópicas" . Revista de Fibrosis Quística . 1 (1): 13-29. doi : 10.1016 / S1569-1993 (01) 00003-0 . PMID 15463806 .
- Tsui LC (1992). "Mutaciones y variaciones de secuencia detectadas en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR): un informe del Consorcio de Análisis Genético de Fibrosis Quística". Mutación humana . 1 (3): 197–203. doi : 10.1002 / humu.1380010304 . PMID 1284534 . S2CID 35904538 .
- McIntosh I, Cutting GR (julio de 1992). "Regulador de la conductancia transmembrana de la fibrosis quística y la etiología y patogénesis de la fibrosis quística". Revista FASEB . 6 (10): 2775–82. doi : 10.1096 / fasebj.6.10.1378801 . PMID 1378801 . S2CID 24932803 .
- Drumm ML, Collins FS (1993). "Biología molecular de la fibrosis quística". Medicina Genética Molecular . 3 : 33–68. doi : 10.1016 / b978-0-12-462003-2.50006-7 . ISBN 9780124620032. PMID 7693108 .
- Kerem B, Kerem E (1996). "La base molecular de la variabilidad de la enfermedad en la fibrosis quística". Revista europea de genética humana . 4 (2): 65–73. doi : 10.1159 / 000472174 . PMID 8744024 . S2CID 41476164 .
- Devidas S, Guggino WB (octubre de 1997). "CFTR: dominios, estructura y función". Revista de Bioenergética y Biomembranas . 29 (5): 443–51. doi : 10.1023 / A: 1022430906284 . PMID 9511929 . S2CID 6000695 .
- Nagel G (diciembre de 1999). "Función diferencial de los dos dominios de unión de nucleótidos en el regulador de conductancia transmembrana de fibrosis quística" . Biochimica et Biophysica Acta . 1461 (2): 263–74. doi : 10.1016 / S0005-2736 (99) 00162-5 . PMID 10581360 .
- Boyle MP (2000). "Presentaciones únicas y complicaciones crónicas en la fibrosis quística del adulto: ¿nos enseñan algo sobre CFTR?" . Investigación respiratoria . 1 (3): 133–5. doi : 10.1186 / rr23 . PMC 59552 . PMID 11667976 .
- Greger R, Schreiber R, Mall M, Wissner A, Hopf A, Briel M, et al. (2001). "Fibrosis quística y CFTR". Pflugers Archiv . 443 Supl. 1: S3-7. doi : 10.1007 / s004240100635 . PMID 11845294 . S2CID 8057614 .
- Bradbury NA (2001). "Cascadas de señalización de AMPc y CFTR: ¿hay más que aprender?". Pflugers Archiv . 443 Suppl 1: S85-91. doi : 10.1007 / s004240100651 . PMID 11845310 . S2CID 19373036 .
- Dahan D, Evagelidis A, Hanrahan JW, Hinkson DA, Jia Y, Luo J, Zhu T (2001). "Regulación del canal CFTR por fosforilación". Pflugers Archiv . 443 Supl. 1: S92-6. doi : 10.1007 / s004240100652 . PMID 11845311 . S2CID 8144727 .
- Cohn JA, Noone PG, Jowell PS (septiembre de 2002). "Pancreatitis idiopática relacionada con CFTR: herencia compleja e identificación de un gen modificador". Revista de Medicina de Investigación . 50 (5): 247S – 255S. doi : 10.1136 / jim-50-suppl5-01 . PMID 12227654 . S2CID 34017638 .
- Schwartz M (febrero de 2003). "[Gen del regulador de la conductancia transmembrana de la fibrosis quística (CFTR): mutaciones y fenotipos clínicos]". Ugeskrift para Laeger . 165 (9): 912–6. PMID 12661515 .
- Wong LJ, Alper OM, Wang BT, Lee MH, Lo SY (julio de 2003). "Dos mutaciones nulas novedosas en un paciente de fibrosis quística de Taiwán y una encuesta de mutaciones CFTR de Asia oriental". Revista Estadounidense de Genética Médica. Parte A . 120A (2): 296–8. doi : 10.1002 / ajmg.a.20039 . PMID 12833420 . S2CID 41060230 .
- Cuppens H, Cassiman JJ (octubre de 2004). "Mutaciones y polimorfismos de CFTR en la infertilidad masculina" . Revista Internacional de Andrología . 27 (5): 251–6. doi : 10.1111 / j.1365-2605.2004.00485.x . PMID 15379964 .
- Cohn JA, Mitchell RM, Jowell PS (marzo de 2005). "El impacto de la fibrosis quística y las mutaciones del gen PSTI / SPINK1 sobre la susceptibilidad a la pancreatitis crónica". Clínicas de Medicina de Laboratorio . 25 (1): 79–100. doi : 10.1016 / j.cll.2004.12.007 . PMID 15749233 .
- Southern KW, Peckham D (2004). "Establecimiento de un diagnóstico de fibrosis quística" . Enfermedad respiratoria crónica . 1 (4): 205–10. doi : 10.1191 / 1479972304cd044rs . PMID 16281647 .
- Kandula L, Whitcomb DC, Lowe ME (junio de 2006). "Problemas genéticos en la pancreatitis pediátrica". Informes actuales de gastroenterología . 8 (3): 248–53. doi : 10.1007 / s11894-006-0083-8 . PMID 16764792 . S2CID 23606613 .
- Marcet B, Boeynaems JM (diciembre de 2006). "Relaciones entre el regulador de la conductancia transmembrana de la fibrosis quística, los nucleótidos extracelulares y la fibrosis quística". Farmacología y terapéutica . 112 (3): 719–32. doi : 10.1016 / j.pharmthera.2006.05.010 . PMID 16828872 .
- Wilschanski M, Durie PR (agosto de 2007). "Patrones de enfermedad gastrointestinal en la edad adulta asociados con mutaciones en el gen CFTR" . Gut . 56 (8): 1153–63. doi : 10.1136 / gut.2004.062786 . PMC 1955522 . PMID 17446304 .
Enlaces externos
- Entrada de GeneReviews / NCBI / NIH / UW sobre trastornos relacionados con CFTR: fibrosis quística (FQ, mucoviscidosis) y ausencia congénita de los vasos deferentes (CAVD)
- La proteína reguladora de la conductancia transmembrana de la fibrosis quística
- La base de datos de mutaciones de genes humanos - Registros CFTR
- Base de datos de mutaciones de fibrosis quística
- Información CFTR del Laboratorio Nacional de Oak Ridge
- CFTR en OMIM (Centro Nacional de Información Biotecnológica)
- Descripción general de toda la información estructural disponible en el PDB para UniProt : P13569 (regulador de conductancia transmembrana de fibrosis quística humana) en el PDBe-KB .
- Descripción general de toda la información estructural disponible en el PDB para UniProt : P26361 (regulador de conductancia transmembrana de fibrosis quística de ratón) en el PDBe-KB .
| vtmiGalería PDB | |
|---|---|
| |