La secuenciación de Sanger es un método de secuenciación de ADN basado en la incorporación selectiva de didesoxinucleótidos que terminan la cadena por la ADN polimerasa durante la replicación del ADN in vitro . [1] [2] Después de ser desarrollado por Frederick Sanger y sus colegas en 1977, se convirtió en el método de secuenciación más utilizado durante aproximadamente 40 años. Fue comercializado por primera vez por Applied Biosystems en 1986. [3] Más recientemente, la secuenciación Sanger de mayor volumen ha sido reemplazada por métodos de secuenciación "Next-Gen" , especialmente para el genoma automatizado a gran escala.análisis. Sin embargo, el método Sanger sigue siendo de amplio uso, para proyectos de menor escala y para la validación de resultados de próxima generación. Todavía tiene la ventaja sobre las tecnologías de secuenciación de lectura corta (como Illumina) en que puede producir lecturas de secuencia de ADN de> 500 nucleótidos .
Método

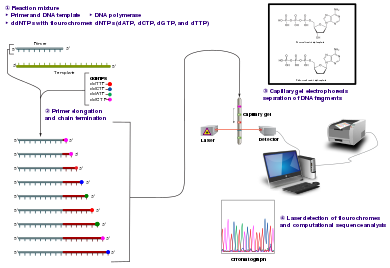
El método clásico de terminación de cadena requiere un molde de ADN monocatenario, un cebador de ADN , una ADN polimerasa , desoxinucleótidos trifosfatos normales (dNTP) y di-desoxinucleótidos trifosfatos modificados (ddNTP), el último de los cuales termina la elongación de la cadena de ADN. Estos nucleótidos que terminan la cadena carecen de un grupo 3'- OH requerido para la formación de un enlace fosfodiéster entre dos nucleótidos, lo que hace que la ADN polimerasa cese la extensión del ADN cuando se incorpora un ddNTP modificado. Los ddNTP se pueden marcar de forma radiactiva o fluorescente para su detección en máquinas de secuenciación automatizadas.
La muestra de ADN se divide en cuatro reacciones de secuenciación separadas, que contienen los cuatro desoxinucleótidos estándar (dATP, dGTP, dCTP y dTTP) y la ADN polimerasa. A cada reacción se le agrega solo uno de los cuatro didesoxinucleótidos (ddATP, ddGTP, ddCTP o ddTTP), mientras que los otros nucleótidos agregados son los ordinarios. La concentración de desoxinucleótido debe ser aproximadamente 100 veces mayor que la del didesoxinucleótido correspondiente (por ejemplo, dTTP 0,5 mM: ddTTP 0,005 mM) para permitir que se produzcan suficientes fragmentos mientras se sigue transcribiendo la secuencia completa (pero la concentración de ddNTP también depende del valor deseado). longitud de la secuencia). [2]Poniéndolo en un orden más sensato, se necesitan cuatro reacciones separadas en este proceso para probar los cuatro ddNTP. Después de rondas de extensión del ADN molde del cebador unido, los fragmentos de ADN resultantes se desnaturalizan por calor y se separan por tamaño usando electroforesis en gel . En la publicación original de 1977, [2] la formación de bucles de ssDNA de pares de bases fue una causa de serias dificultades para resolver bandas en algunos lugares. Esto se realiza frecuentemente usando un gel de poliacrilamida- urea desnaturalizante con cada una de las cuatro reacciones desarrolladas en uno de los cuatro carriles individuales (carriles A, T, G, C). Las bandas de ADN se pueden visualizar mediante autorradiografía o luz ultravioleta y la secuencia de ADN se puede leer directamente en elPelícula de rayos X o imagen de gel.

En la imagen de la derecha, se expuso una película de rayos X al gel y las bandas oscuras corresponden a fragmentos de ADN de diferentes longitudes. Una banda oscura en un carril indica un fragmento de ADN que es el resultado de la terminación de la cadena después de la incorporación de un didesoxinucleótido (ddATP, ddGTP, ddCTP o ddTTP). Las posiciones relativas de las diferentes bandas entre los cuatro carriles, de abajo hacia arriba, se utilizan para leer la secuencia de ADN.

Las variaciones técnicas de la secuenciación de terminación de cadena incluyen el marcado con nucleótidos que contienen fósforo radiactivo para el marcaje radiactivo , o el uso de un cebador marcado en el extremo 5 'con un tinte fluorescente . La secuenciación de imprimación de tinte facilita la lectura en un sistema óptico para un análisis y automatización más rápidos y económicos. El desarrollo posterior de Leroy Hood y colaboradores [4] [5] de cebadores y ddNTP marcados con fluorescencia preparó el escenario para la secuenciación de ADN automatizada de alto rendimiento.

Los métodos de terminación de cadena han simplificado enormemente la secuenciación del ADN. Por ejemplo, los kits basados en terminación de cadena están disponibles comercialmente que contienen los reactivos necesarios para la secuenciación, pre-alícuotas y listos para usar. Las limitaciones incluyen la unión no específica del cebador al ADN, que afecta la lectura precisa de la secuencia de ADN, y las estructuras secundarias del ADN que afectan la fidelidad de la secuencia.
Secuenciación del terminador de tinte

La secuenciación del terminador de colorante utiliza el marcaje del terminador de cadena ddNTP, que permite la secuenciación en una sola reacción, en lugar de cuatro reacciones como en el método de cebador marcado. En la secuenciación del terminador de colorante, cada uno de los cuatro terminadores de cadena de didesoxinucleótidos se marca con colorantes fluorescentes, cada uno de los cuales emite luz en diferentes longitudes de onda .
Debido a su mayor conveniencia y velocidad, la secuenciación del terminador de tinte es ahora el pilar de la secuenciación automatizada. Sus limitaciones incluyen efectos de colorante debido a diferencias en la incorporación de los terminadores de cadena marcados con colorante en el fragmento de ADN, lo que resulta en alturas y formas de pico desiguales en el cromatograma de trazas de secuencia de ADN electrónico después de la electroforesis capilar (ver figura a la izquierda).
Este problema se ha abordado con el uso de colorantes y sistemas enzimáticos de ADN polimerasa modificados que minimizan la variabilidad de la incorporación, así como con métodos para eliminar las "manchas de colorante". El método de secuenciación por tinte-terminador, junto con los analizadores automáticos de secuencias de ADN de alto rendimiento, se utilizó para la gran mayoría de proyectos de secuenciación hasta la introducción de la secuenciación de próxima generación .
Automatización y preparación de muestras

Los instrumentos de secuenciación de ADN automatizados ( secuenciadores de ADN ) pueden secuenciar hasta 384 muestras de ADN en un solo lote. Las ejecuciones por lotes pueden ocurrir hasta 24 veces al día. Los secuenciadores de ADN separan las hebras por tamaño (o longitud) mediante electroforesis capilar , detectan y registran la fluorescencia del tinte y emiten datos como cromatogramas de trazas de picos fluorescentes . Reacciones de secuenciación ( termociclado y etiquetado), limpieza y resuspensión de muestras en una solución tampónse realizan por separado, antes de cargar las muestras en el secuenciador. Varios paquetes de software comerciales y no comerciales pueden recortar automáticamente los rastros de ADN de baja calidad. Estos programas puntúan la calidad de cada pico y eliminan los picos base de baja calidad (que generalmente se encuentran en los extremos de la secuencia). La precisión de tales algoritmos es inferior a la del examen visual por un operador humano, pero es adecuada para el procesamiento automatizado de grandes conjuntos de datos de secuencia.
Desafíos
Los desafíos comunes de la secuenciación de ADN con el método Sanger incluyen la mala calidad en las primeras 15-40 bases de la secuencia debido a la unión del cebador y el deterioro de la calidad de las trazas de secuenciación después de 700-900 bases. El software de llamadas de base, como Phred, generalmente proporciona una estimación de la calidad para ayudar a recortar regiones de secuencias de baja calidad. [6] [7]
En los casos en los que se clonan fragmentos de ADN antes de la secuenciación, la secuencia resultante puede contener partes del vector de clonación . Por el contrario, la clonación basada en PCR y las tecnologías de secuenciación de próxima generación basadas en la pirosecuenciación a menudo evitan el uso de vectores de clonación. Recientemente, se han desarrollado métodos de secuenciación de Sanger en un solo paso (amplificación y secuenciación combinadas) como Ampliseq y SeqSharp que permiten la secuenciación rápida de genes diana sin clonación o amplificación previa. [8] [9]
Los métodos actuales pueden secuenciar directamente solo fragmentos de ADN relativamente cortos (300-1000 nucleótidos de longitud) en una sola reacción. El principal obstáculo para la secuenciación de fragmentos de ADN por encima de este límite de tamaño es el poder de separación insuficiente para resolver grandes fragmentos de ADN que difieren en longitud en un solo nucleótido.
Secuenciación microfluídica de Sanger
La secuenciación microfluídica de Sanger es una aplicación de laboratorio en un chip para la secuenciación de ADN, en la que los pasos de secuenciación de Sanger (ciclos térmicos, purificación de muestras y electroforesis capilar) se integran en un chip a escala de oblea utilizando volúmenes de muestra a escala de nanolitros. Esta tecnología genera lecturas de secuencia largas y precisas, al tiempo que elimina muchas de las deficiencias significativas del método Sanger convencional (por ejemplo, alto consumo de reactivos costosos, dependencia de equipos costosos, manipulaciones intensivas en personal, etc.) al integrar y automatizar los pasos de secuenciación de Sanger. .
En sus inicios modernos, la secuenciación del genoma de alto rendimiento implica la fragmentación del genoma en pequeñas piezas monocatenarias, seguido de la amplificación de los fragmentos mediante la reacción en cadena de la polimerasa (PCR). Adoptando el método de Sanger, cada fragmento de ADN se termina irreversiblemente con la incorporación de un nucleótido terminal de cadena didesoxi marcado con fluorescencia, produciendo así una "escalera" de ADN de fragmentos que difieren en longitud en una base y llevan una etiqueta fluorescente específica de base en la base del terminal. Las escaleras de base amplificadas se separan luego mediante electroforesis de matriz capilar (CAE) con automatización in situDetección de "línea de meta" de los fragmentos de ssDNA marcados con fluorescencia, que proporciona una secuencia ordenada de los fragmentos. Estas lecturas de secuencia se ensamblan luego por computadora en secuencias superpuestas o contiguas (denominadas "contigs") que se asemejan a la secuencia genómica completa una vez ensambladas por completo. [10]
Los métodos de Sanger alcanzan longitudes de lectura de aproximadamente 800 pb (normalmente 500-600 pb con ADN no enriquecido). Las longitudes de lectura más largas en los métodos de Sanger muestran ventajas significativas sobre otros métodos de secuenciación, especialmente en términos de secuenciar regiones repetitivas del genoma. Un desafío de los datos de secuencia de lectura corta es particularmente un problema en la secuenciación de nuevos genomas (de novo) y en la secuenciación de segmentos genómicos altamente reorganizados, típicamente los que se ven en los genomas del cáncer o en regiones de cromosomas que exhiben variación estructural. [11]
Aplicaciones de las tecnologías de secuenciación de microfluidos
Otras aplicaciones útiles de la secuenciación del ADN incluyen polimorfismo de un solo nucleótido (SNP) de detección, -polimorfismo de conformación monocatenaria (SSCP) análisis de heterodúplex , y repetición corta en tándem de análisis (STR). La resolución de fragmentos de ADN de acuerdo con las diferencias de tamaño y / o conformación es el paso más crítico en el estudio de estas características del genoma. [10]
Diseño de dispositivo
El chip de secuenciación tiene una construcción de cuatro capas, que consta de tres obleas de vidrio de 100 mm de diámetro (en las que se microfabrican los elementos del dispositivo) y una membrana de polidimetilsiloxano (PDMS). Las cámaras de reacción y los canales de electroforesis capilar están grabados entre las dos obleas de vidrio superiores, que están unidas térmicamente. Las interconexiones de canales tridimensionales y las microválvulas están formadas por el PDMS y la oblea de vidrio del colector inferior.
El dispositivo consta de tres unidades funcionales, cada una de las cuales corresponde a los pasos de secuenciación de Sanger. La unidad de ciclo térmico (TC) es una cámara de reacción de 250 nanolitros con detector de temperatura resistivo integrado, microválvulas y un calentador de superficie. El movimiento del reactivo entre la capa superior totalmente de vidrio y la capa inferior de vidrio-PDMS se produce a través de orificios pasantes de 500 μm de diámetro. Después del ciclo térmico, la mezcla de reacción se purifica en la cámara de captura / purificación y luego se inyecta en la cámara de electroforesis capilar (CE). La unidad CE consta de un capilar de 30 cm que se pliega en un patrón de conmutación compacto mediante giros de 65 μm de ancho.
Química de secuenciación
- Ciclos térmicos
- En la cámara de reacción de TC, el reactivo de secuenciación del terminador de tinte, el ADN molde y los cebadores se cargan en la cámara de TC y se someten a un ciclo térmico durante 35 ciclos (a 95 ° C durante 12 segundos y a 60 ° C durante 55 segundos).
- Purificación
- La mezcla de reacción cargada (que contiene fragmentos de extensión, ADN molde y reactivo de secuenciación en exceso) se conduce a través de una cámara de captura / purificación a 30 ° C mediante un campo eléctrico de 33 voltios / cm aplicado entre la salida de captura y los puertos de entrada. El gel de captura a través del cual se conduce la muestra consiste en 40 μM de oligonucleótido (complementario a los cebadores) unido covalentemente a una matriz de poliacrilamida. Los fragmentos de extensión son inmovilizados por la matriz de gel y el cebador en exceso, la plantilla, los nucleótidos libres y las sales se eluyen a través del puerto de desechos de captura. El gel de captura se calienta a 67-75 ° C para liberar fragmentos de extensión.
- Electroforesis capilar
- Los fragmentos de extensión se inyectan en la cámara de CE donde se someten a electroforesis a través de un campo de 125-167 V / cm.
Plataformas
La plataforma Apollo 100 (Microchip Biotechnologies Inc., Dublin, CA) [12] integra los dos primeros pasos de secuenciación de Sanger (ciclos térmicos y purificación) en un sistema totalmente automatizado. El fabricante afirma que las muestras están listas para la electroforesis capilar dentro de las tres horas posteriores a la carga de la muestra y los reactivos en el sistema. La plataforma Apollo 100 requiere volúmenes de reactivos por debajo de los microlitros.
Comparaciones con otras técnicas de secuenciación
| Tecnología | Numero de carriles | Volumen de inyección (nL) | Tiempo de análisis | Longitud promedio de lectura | Rendimiento (incluido el análisis; Mb / h ) | Verter gel | Seguimiento de carril |
|---|---|---|---|---|---|---|---|
| Gel de losa | 96 | 500–1000 | 6 a 8 horas | 700 pb | 0.0672 | sí | sí |
| Electroforesis de matriz capilar | 96 | 1–5 | 1-3 horas | 700 pb | 0,166 | No | No |
| Pastilla | 96 | 0,1-0,5 | 6 a 30 minutos | 430 pb | 0,660 | No | No |
| 454 / Roche FLX (2008) | <0,001 | 4 horas | 200–300 pb | 20-30 | |||
| Illumina / Solexa (2008) | 2-3 días | 30-100 pb | 20 | ||||
| ABI / SOLiD (2008) | 8 dias | 35 pb | 5-15 | ||||
| Illumina MiSeq (2019) | 1-3 días | 2x75–2x300 pb | 170–250 | ||||
| Illumina NovaSeq (2019) | 1-2 días | 2x50–2x150 pb | 22.000–67.000 | ||||
| Ion Torrent Ion 530 (2019) | 2,5–4 horas | 200–600 pb | 110–920 | ||||
| BGI MGISEQ-T7 (2019) | 1 día | 2x150 pb | 250.000 | ||||
| Pacific Biosciences SMRT (2019) | 10-20 horas | 10–30 KB | 1300 | ||||
| Oxford Nanopore MinIon (2019) | 3 días | 13-20 KB [15] | 700 |
El objetivo final de la secuenciación de alto rendimiento es desarrollar sistemas que sean de bajo costo y extremadamente eficientes para obtener longitudes de lectura extendidas (más largas). Las longitudes de lectura más largas de cada separación electroforética individual reducen sustancialmente el costo asociado con la secuenciación de ADN de novo y el número de plantillas necesarias para secuenciar los contigs de ADN con una redundancia dada. Los microfluidos pueden permitir un ensamblaje de secuencia más rápido, económico y sencillo. [10]
Referencias
- ^ Sanger F; Coulson AR (mayo de 1975). "Un método rápido para la determinación de secuencias de ADN mediante síntesis cebada con ADN polimerasa". J. Mol. Biol . 94 (3): 441–8. doi : 10.1016 / 0022-2836 (75) 90213-2 . PMID 1100841 .
- ^ a b c Sanger F; Nicklen S; Coulson AR (diciembre de 1977). "Secuenciación de ADN con inhibidores de terminación de cadena" . Proc. Natl. Acad. Sci. USA . 74 (12): 5463–7. Código bibliográfico : 1977PNAS ... 74.5463S . doi : 10.1073 / pnas.74.12.5463 . PMC 431765 . PMID 271968 .
- ^ Adams, Jill U. (2008). "Tecnologías de secuenciación de ADN" . Educación en la naturaleza . Consultado el 24 de octubre de 2019 .
- ^ Smith LM, Sanders JZ, Kaiser RJ, et al. (1986). "Detección de fluorescencia en análisis automatizado de secuencias de ADN". Naturaleza . 321 (6071): 674–9. Código Bibliográfico : 1986Natur.321..674S . doi : 10.1038 / 321674a0 . PMID 3713851 . S2CID 27800972 .
Hemos desarrollado un método para la automatización parcial del análisis de secuencias de ADN. La detección por fluorescencia de los fragmentos de ADN se logra mediante un fluoróforo unido covalentemente al cebador oligonucleotídico utilizado en el análisis enzimático de la secuencia de ADN. Se usa un fluoróforo de diferente color para cada una de las reacciones específicas para las bases A, C, G y T. Las mezclas de reacción se combinan y se someten a una electroforesis conjunta en un solo tubo de gel de poliacrilamida, las bandas fluorescentes de ADN separadas se detectan cerca del fondo del tubo, y la información de secuencia se adquiere directamente por computadora.
- ^ Smith LM; Fung S; Hunkapiller MW; Hunkapiller TJ; Hood LE (abril de 1985). "La síntesis de oligonucleótidos que contienen un grupo amino alifático en el extremo 5 ': síntesis de cebadores de ADN fluorescentes para su uso en el análisis de secuencias de ADN" . Ácidos nucleicos Res . 13 (7): 2399–412. doi : 10.1093 / nar / 13.7.2399 . PMC 341163 . PMID 4000959 .
- ^ "Phred - Base de llamadas de calidad" . Consultado el 24 de febrero de 2011 .
- ^ Ledergerber, C; Dessimoz, C (2011). "Bases de llamadas para plataformas de secuenciación de próxima generación" . Sesiones informativas en bioinformática . 12 (5): 489–97. doi : 10.1093 / bib / bbq077 . PMC 3178052 . PMID 21245079 .
- ^ Murphy, K .; Berg, K .; Eshleman, J. (2005). "Secuenciación de ADN genómico mediante amplificación combinada y reacción de secuenciación cíclica" . Química clínica . 51 (1): 35–39. doi : 10.1373 / clinchem.2004.039164 . PMID 15514094 .
- ^ Sengupta, D..; Cookson, B.. (2010). "SeqSharp: un enfoque general para mejorar la secuenciación del ciclo que facilita un método robusto de secuenciación y amplificación combinada de un solo paso" . La Revista de Diagnóstico Molecular . 12 (3): 272–277. doi : 10.2353 / jmoldx.2010.090134 . PMC 2860461 . PMID 20203000 .
- ^ a b c Kan, Cheuk-Wai; Fredlake, Christopher P .; Doherty, Erin AS; Barron, Annelise E. (1 de noviembre de 2004). "Secuenciación de ADN y genotipado en sistemas de electroforesis miniaturizados". Electroforesis . 25 (21-22): 3564-3588. doi : 10.1002 / elps.200406161 . PMID 15565709 . S2CID 4851728 .
- ^ a b Morozova, Olena; Marra, Marco A (2008). "Aplicaciones de las tecnologías de secuenciación de última generación en genómica funcional" . Genómica . 92 (5): 255–64. doi : 10.1016 / j.ygeno.2008.07.001 . PMID 18703132 .
- ^ Microchip Biologies Inc. Apollo 100
- ^ Sinville, Rondedrick; Soper, Steven A (2007). "Separaciones de ADN de alta resolución mediante electroforesis en microchip". Revista de ciencia de la separación . 30 (11): 1714–28. doi : 10.1002 / jssc.200700150 . PMID 17623451 .
- ^ Kumar, Kishore; Cowley, Mark; Davis, Ryan (2019). "Secuenciación de próxima generación y tecnologías emergentes" . Seminarios de Trombosis y Hemostasia . 45 (7): 661–673. doi : 10.1055 / s-0039-1688446 . ISSN 0094-6176 . PMID 31096307 .
- ^ Tyson, John R .; O'Neil, Nigel J .; Jain, Miten; Olsen, Hugh E .; Hieter, Philip; Snutch, Terrance P. (2018). "La secuenciación y el ensamblaje de lectura larga basada en MinION amplía el genoma de referencia de Caenorhabditis elegans" . Investigación del genoma . 28 (2): 266-274. doi : 10.1101 / gr.221184.117 . ISSN 1088-9051 . PMC 5793790 . PMID 29273626 .
Lectura adicional
- Dewey, F. E; Pan, S; Wheeler, M. T; Quake, S. R; Ashley, E. A (2012). "Secuenciación de ADN: aplicaciones clínicas de nuevas tecnologías de secuenciación de ADN" . Circulación . 125 (7): 931–44. doi : 10.1161 / CIRCULATIONAHA.110.972828 . PMC 3364518 . PMID 22354974 .
- Sanger, F; Coulson, AR; Barrell, BG; Smith, AJH; Roe, BA (1980). "Clonación en bacteriófagos monocatenarios como ayuda para la secuenciación rápida del ADN". Revista de Biología Molecular . 143 (2): 161–78. doi : 10.1016 / 0022-2836 (80) 90196-5 . PMID 6260957 .
Enlaces externos
- MBI dice que la nueva herramienta que automatiza la preparación de muestras de Sanger reduce los costos de reactivos y mano de obra