Ictiosis ligada al cromosoma X
| Ictiosis ligada al cromosoma X | |
|---|---|
| Otros nombres | Deficiencia de esteroides sulfatasa, ictiosis recesiva ligada al cromosoma X [1] |
 | |
| Herencia recesiva ligada al cromosoma X: los niños afectados pueden heredar una deleción o mutación del gen STS de sus madres | |
| Especialidad | Genética Médica |
La ictiosis ligada al cromosoma X (abreviado XLI ) es una afección de la piel causada por la deficiencia hereditaria de la enzima esteroide sulfatasa (STS) que afecta a 1 de cada 2000 a 1 de cada 6000 hombres. [2] XLI se manifiesta con piel seca y escamosa [3] y se debe a deleciones [4] [5] o mutaciones [6] en el gen STS . XLI también puede ocurrir en el contexto de deleciones más grandes que causan síndromes de genes contiguos . [4] El tratamiento está dirigido principalmente a aliviar los síntomas de la piel. [7] El término proviene del griego antiguo 'ichthys' que significa 'pez'.
Signos y síntomas

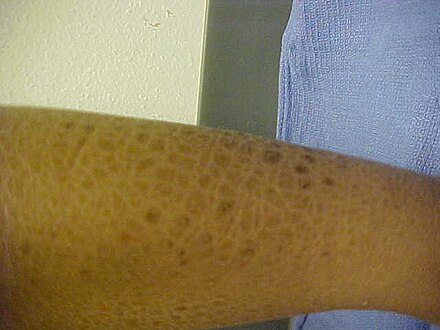
Los síntomas principales de XLI incluyen descamación de la piel, particularmente en el cuello, el tronco y las extremidades inferiores. Las superficies extensoras suelen ser las áreas más gravemente afectadas. Las escamas de> 4 mm de diámetro se adhieren a la piel subyacente y pueden ser de color marrón oscuro o gris. Los síntomas pueden desaparecer durante el verano. [2]
Condiciones médicas asociadas
Aparte de la descamación de la piel, XLI no suele asociarse con otros problemas médicos importantes. [8] La fibrilación auricular o el aleteo auricular pueden afectar hasta 1 de cada 10 hombres con XLI. [9] Puede haber opacidades corneales, pero no afectan la visión. Se informa criptorquidia en algunas personas. [2] Las personas con XLI parecen tener un mayor riesgo de trastornos del desarrollo como el autismo y el trastorno por déficit de atención con hiperactividad y algunas personas afectadas presentan problemas de estado de ánimo [10] Las personas con XLI pueden presentar discapacidad intelectual, aunque se cree que esto se debe a deleciones que abarcan genes vecinos además de STS . [11]
Las mujeres portadoras generalmente no experimentan ninguno de estos problemas, pero pueden tener dificultades durante el parto, ya que el STS expresado en la placenta juega un papel en el trabajo de parto normal. [12] Las mujeres portadoras también pueden tener un riesgo ligeramente mayor de desarrollar problemas de salud mental después del parto [13]. Por estas razones, las portadoras deben asegurarse de que su obstetra esté al tanto de la afección. [ cita requerida ]
Genética
{kind=link}
{kind=link}
El gen STS se encuentra en el cromosoma X en la banda Xp22.3. Por lo tanto, el síndrome es una afección ligada al cromosoma X y afecta a hombres y mujeres de manera diferente. El par 23 de cromosomas se denomina típicamente "cromosomas sexuales". Las mujeres tienen dos cromosomas X y los hombres tienen un cromosoma X y uno Y. Por lo tanto, en individuos normales, los machos portan una sola copia del gen STS y las hembras portan dos copias. Este gen escapa parcialmente a la inactivación de X y las hembras normalmente expresan mayores cantidades de la enzima STS que los machos. [14]
XLI puede ocurrir a través de nuevas deleciones o mutaciones del gen STS , pero es más común que se herede de una madre portadora. [15] Una deleción o mutación hemicigótica del gen STS en un hombre da como resultado una ausencia completa de actividad enzimática, mientras que una mujer portadora de una mutación o deleción es heterocigótica y aún tiene una copia normal del gen STS . Las mujeres portadoras de una deleción o mutación de STS todavía expresan la enzima STS, aunque con una actividad enzimática disminuida. [dieciséis]
Por esta razón, XLI afecta con mayor frecuencia a los hombres, aunque las personas con anomalías numéricas de los cromosomas sexuales (45, X y 47, XXY) que también portan deleciones o mutaciones de STS serían excepciones a esta regla. [ cita requerida ]
Además, una mujer podría verse afectada si fuera la descendencia de un macho afectado y una hembra portadora y heredara una deleción o mutación del gen STS en ambos cromosomas X.
Problemas de asesoramiento genético
Dado que la mayoría de los casos parecen ocurrir a través de la transmisión de una deleción de STS de una madre portadora, [15] deben realizarse pruebas enzimáticas o pruebas de ADN en la madre de cualquier caso simplex recién diagnosticado (es decir, el primer caso en una familia). En el caso de una familia extensa con muchas personas afectadas, el estado de portador a menudo se puede asignar en función del análisis del pedigrí.
- Los machos con XLI transmitirán el cromosoma X que alberga la deleción o mutación de STS a cada una de sus crías hembras, que por lo tanto serán portadoras obligatorias . Sin embargo, todos los descendientes masculinos no se verán afectados, ya que reciben el cromosoma Y de su padre.
- Las mujeres portadoras de una deleción o mutación de STS tienen un 50% de probabilidad con cada embarazo de transmitirlo a una descendencia. Por lo tanto, cada descendiente masculino tiene un 50% de posibilidades de verse afectado por XLI, mientras que cada descendiente hembra tiene un 50% de posibilidades de ser portador de esta afección. Cualquier individuo que herede la copia normal del gen STS de la madre no se verá afectado y tendrá una probabilidad extremadamente baja de tener un hijo afectado con esta afección.
Debido a la segregación aleatoria de los cromosomas durante la gametogénesis , cada embarazo estará sujeto a las mismas probabilidades, independientemente del número de descendientes previamente afectados o no afectados. Los riesgos de recurrencia anteriores se basan en la suposición de que un hombre afectado o una mujer portadora tendrá hijos con un individuo no afectado o no portador. Los riesgos de tener descendencia afectada aumentarían claramente en el caso de una unión entre un macho con XLI y una hembra portadora.
Fisiología / bioquímica
La enzima STS (EC 3.1.6.2), también conocida como arilsulfatasa C, se expresa en todo el cuerpo, con mayor expresión en la piel, hígado, ganglios linfáticos y placenta, y menor expresión en tejido mamario y cerebro [17] STS cataliza la hidrólisis de esteroides sulfatados, como el sulfato de estrona y el sulfato de dehidroepiandrosterona (DHEAS), a esteroides no sulfatados estradiol y androstenediol, respectivamente. [18] Prenatalmente, la enzima participa en la producción de estrógenos placentarios . [19] La enzima también participa en la producción de esteroides suprarrenales, así como en la conversión de esteroides sulfatados en otros tejidos. [ cita requerida ]
Parece haber un papel particularmente importante para la enzima en la piel. La deficiencia de la enzima conduce a la característica piel seca y escamosa que se observa en la ictiosis. Investigaciones recientes indican que las anomalías cutáneas observadas en XLI pueden deberse a la acumulación de sulfato de colesterol en la epidermis externa, lo que conduce a una función de barrera anormal y retención de corneocitos. [20]
Diagnóstico
Se puede sospechar de XLI en función de los hallazgos clínicos, aunque los síntomas pueden tardar un tiempo variable en manifestarse, desde unas pocas horas después del nacimiento, hasta un año en los casos más leves. El diagnóstico generalmente lo realiza un dermatólogo , que también suele formular el plan de tratamiento (ver más abajo). La deficiencia de la enzima STS se confirma mediante un ensayo bioquímico clínicamente disponible. La detección de portadores se puede realizar en madres de hijos afectados mediante esta prueba (consulte Genética, a continuación). [16] También se ofrecen pruebas moleculares para detectar deleciones o mutaciones del ADN y pueden ser particularmente útiles en la evaluación de personas con afecciones médicas asociadas (ver más abajo). Diagnóstico prenatales posible utilizando pruebas bioquímicas o moleculares. Sin embargo, el uso del diagnóstico prenatal para afecciones genéticas que se consideran generalmente benignas plantea serias consideraciones éticas y requiere un asesoramiento genético detallado. [ cita requerida ]
Tratamiento
Debido a que la XLI es causada por una mutación o deleción genética, no existe una "cura". Uno de los objetivos del tratamiento es reducir la descamación eliminando el exceso de escamas escamosas y mantener la piel hidratada. Esto se puede lograr usando una variedad de cremas tópicas. [2] [7]
- Los agentes queratolíticos como el lactato de amonio (Lac-Hydrin) se utilizan para facilitar la liberación de los corneocitos retenidos.
- Isotretinoína tópica
- El tazaroteno retinoide selectivo de receptor tópico [21]
Se están realizando investigaciones con respecto al uso de la terapia génica para tratar la XLI. [22]
Historia
En la década de 1960, la ictiosis recesiva ligada al cromosoma X se distinguía clínicamente de otras ictiosis. [23] : 486 [24] : 561
Ver también
- Ictiosis
- Síndrome de carvajal
Referencias
- ^ Rapini, Ronald P .; Bolognia, Jean L .; Jorizzo, Joseph L. (2007). Dermatología: Set de 2 volúmenes . San Luis: Mosby. ISBN 978-1-4160-2999-1.
- ^ a b c d Carlo Gelmetti; Caputo, Ruggero (2002). Dermatología y dermatopatología pediátricas: un atlas conciso . T&F STM. pag. 160. ISBN 978-1-84184-120-5.
- ^ Herencia mendeliana en línea en el hombre (OMIM): ICHTHYOSIS, X-LINKED - 308100
- ^ a b Ballabio A, Parenti G, Carrozzo R, et al. (1987). "Aislamiento y caracterización de un clon de ADNc de esteroide sulfatasa: deleciones genómicas en pacientes con ictiosis ligada al cromosoma X" . Proc. Natl. Acad. Sci. USA . 84 (13): 4519–23. Código Bibliográfico : 1987PNAS ... 84.4519B . doi : 10.1073 / pnas.84.13.4519 . PMC 305121 . PMID 3474618 .
- ^ Bonifas JM, Morley BJ, Oakey RE, Kan YW, Epstein EH (diciembre de 1987). "Clonación de un ADNc para esteroide sulfatasa: ocurrencia frecuente de deleciones de genes en pacientes con ictiosis recesiva ligada al cromosoma X" . Proc. Natl. Acad. Sci. USA . 84 (24): 9248–51. Código Bibliográfico : 1987PNAS ... 84.9248B . doi : 10.1073 / pnas.84.24.9248 . PMC 299730 . PMID 3480541 .
- ^ Basler E, Grompe M, Parenti G, Yates J, Ballabio A (marzo de 1992). "Identificación de mutaciones puntuales en el gen de la esteroide sulfatasa de tres pacientes con ictiosis ligada al cromosoma X" . Soy. J. Hum. Genet . 50 (3): 483–91. PMC 1684279 . PMID 1539590 .
- ^ a b Ictiosis ligada al cromosoma X en eMedicine : Sección de tratamiento
- ^ DiGiovanna JJ, Robinson-Bostom L (2003). "Ictiosis: etiología, diagnóstico y manejo". Soy J Clin Dermatol . 4 (2): 81–95. doi : 10.2165 / 00128071-200304020-00002 . PMID 12553849 .
- ^ Brcic, Lucija; Underwood, Jack FG; Kendall, Kimberley M .; Caseras, Xavier; Kirov, George; Davies, William (2020). "Fenotipos médicos y neuroconductuales en portadores de deleciones genéticas asociadas a ictiosis ligada al cromosoma X en el Biobanco del Reino Unido" . Revista de Genética Médica . 57 (10): 692–698. doi : 10.1136 / jmedgenet-2019-106676 . PMC 7525778 . PMID 32139392 .
- ^ Chatterjee S, Humby T, Davies W (2016) Fenotipos conductuales y psiquiátricos en hombres y niños con ictiosis ligada al cromosoma X: evidencia de una encuesta mundial en línea. PLoS One 11 (10): e0164417 PMID: 27711218 doi: 10.1371 / journal.pone.0164417 URL: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0164417
- ^ Van Esch H, holandeses K, Badisco L, et al. (2005). "La eliminación de VCX-A debido a NAHR juega un papel importante en la aparición de retraso mental en pacientes con ictiosis ligada al cromosoma X" . Tararear. Mol. Genet . 14 (13): 1795–803. doi : 10.1093 / hmg / ddi186 . PMID 15888481 .
- ^ Bradshaw KD, Carr BR (1986). "Deficiencia placentaria de sulfatasa: expresión materna y fetal de la deficiencia de esteroides sulfatasa e ictiosis ligada al cromosoma X". Obstet Gynecol Surv . 41 (7): 401-13. PMID 3531932 .
- ^ Cavenagh A, Chatterjee S, Davies W (2019) Fenotipos conductuales y psiquiátricos en mujeres portadoras de mutaciones genéticas asociadas con ictiosis ligada al cromosoma X. PLoS One 14 (2): e0212330 PMID: 30768640 doi: 10.1371 / journal.pone.0212330 URL: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0212330
- ^ Lykkesfeldt G, Lykkesfeldt AE, Skakkebaek NE (1984). "Esteroide sulfatasa en el hombre: un locus X no inactivado con compensación de dosis de genes parcial". Tararear. Genet . 65 (4): 355–7. doi : 10.1007 / BF00291559 . PMID 6582028 . S2CID 2625156 .
- ^ a b Valdes-Flores M, Kofman-Alfaro SH, Jimenez-Vaca AL, Cuevas-Covarrubias SA (agosto de 2001). "Identificación de portadores por análisis FISH en casos aislados de ictiosis ligada al cromosoma X". Soy. J. Med. Genet . 102 (2): 146–8. doi : 10.1002 / ajmg.1450 . PMID 11477606 .
- ↑ a b Cuevas-Covarrubias SA, Kofman-Alfaro S, Orozco Orozco E, Diaz-Zagoya JC (1995). "La identificación bioquímica del estado de portador en madres de casos esporádicos de ictiosis recesiva ligada al cromosoma X". Gineta. Condes . 6 (2): 103–7. PMID 7546451 .
- ^ Selcer KW, Difrancesca HM, Chandra AB, Li PK (2007). "Análisis inmunohistoquímico de esteroides sulfatasa en tejidos humanos". J. Steroid Biochem. Mol. Biol . 105 (1–5): 115–23. doi : 10.1016 / j.jsbmb.2006.12.105 . PMID 17604157 . S2CID 22124602 .
- ^ Reed MJ, Purohit A, Woo LW, Newman SP, Potter BV (abril de 2005). "Esteroide sulfatasa: biología molecular, regulación e inhibición" . Endocr. Rev . 26 (2): 171–202. doi : 10.1210 / er.2004-0003 . PMID 15561802 .
- ^ Jöbsis AC, De Groot WP, Tigges AJ, et al. (1980). "Ictiosis ligada al cromosoma X y deficiencia de sulfatasa placentaria ligada al cromosoma X: una entidad patológica. Observaciones histoquímicas" . Soy. J. Pathol . 99 (2): 279–89. PMC 1903491 . PMID 6929654 .
- ^ Elias PM, Crumrine D, Rassner U, et al. (2004). "Base para la disfunción de la barrera de descamación y permeabilidad anormal en RXLI" . J. Invest. Dermatol . 122 (2): 314–9. doi : 10.1046 / j.1523-1747.2003.22258.x . PMID 15009711 .
- ^ Cotellessa C, Cuevas-Covarrubias SA, Valeri P, Fargnoli MC, Peris K (2005). "Tazaroteno tópico al 0.05% versus tratamiento con ácido glicólico al 70% en la ictiosis ligada al cromosoma X debido a una extensa deleción del gen STS" . Acta Derm. Venereol . 85 (4): 346–8. doi : 10.1080 / 00015550510026613 . PMID 16191859 .
- ^ Freiberg RA, Choate KA, Deng H, Alperin ES, Shapiro LJ, Khavari PA (1997). "Un modelo de transferencia genética correctiva en la ictiosis ligada al cromosoma X" . Tararear. Mol. Genet . 6 (6): 927–33. doi : 10.1093 / hmg / 6.6.927 . PMID 9175741 .
- ^ Freedberg, et al. (2003). Dermatología de Fitzpatrick en Medicina General . (6ª ed.). McGraw-Hill. ISBN 0-07-138076-0 .
- ^ James, William; Berger, Timothy; Elston, Dirk (2005). Enfermedades de la piel de Andrews: Dermatología clínica . (10ª ed.). Saunders. ISBN 0-7216-2921-0 .
enlaces externos
| Clasificación | D
|
|---|---|
| Recursos externos |
|
- Genodermatosis
- Trastornos del metabolismo del colesterol y los esteroides
- Enfermedades raras