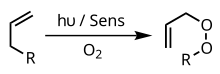
Una fotooxigenación es una reacción de oxidación inducida por la luz en la que se incorpora oxígeno molecular al producto o productos. [1] [2] El interés de la investigación inicial en las reacciones de fotooxigenación surgió de las observaciones de Oscar Raab en 1900 de que la combinación de luz, oxígeno y fotosensibilizadores es altamente tóxica para las células. [3] Los primeros estudios de fotooxigenación se centraron en el daño oxidativo del ADN y los aminoácidos, [2] pero investigaciones recientes han llevado a la aplicación de la fotooxigenación en la síntesis orgánica y la terapia fotodinámica . [4]
Las reacciones de fotooxigenación son iniciadas por un fotosensibilizador , que es una molécula que entra en un estado excitado cuando se expone a la luz de una longitud de onda específica (por ejemplo, tintes y pigmentos). El sensibilizador excitado luego reacciona con un sustrato o con oxígeno molecular en estado fundamental, iniciando una cascada de transferencias de energía que finalmente dan como resultado una molécula oxigenada. En consecuencia, las reacciones de fotooxigenación se clasifican según el tipo y el orden de estos intermediarios (como reacciones de tipo I, tipo II o tipo III [5] ). [2] [3]
Antecedentes
Terminología
Las reacciones de fotooxigenación se confunden fácilmente con varios procesos que muestran nombres similares (es decir, oxidación fotosensibilizada). Se pueden hacer claras distinciones basadas en tres atributos: oxidación , la participación de la luz y la incorporación de oxígeno molecular en los productos:

Sensibilizadores

Los sensibilizadores (denominados "Sens") son compuestos, como tintes de fluoresceína , azul de metileno e hidrocarburos aromáticos policíclicos , que pueden absorber la radiación electromagnética (generalmente en el rango visible del espectro) y eventualmente transferir esa energía al oxígeno molecular o al sustrato del proceso de fotooxigenación. Muchos sensibilizadores, tanto naturales como sintéticos, dependen de amplios sistemas aromáticos para absorber la luz en el espectro visible. [4] Cuando los sensibilizadores se excitan con la luz, alcanzan un estado singlete , 1 Sens *. Este singlete luego se convierte en un estado triplete(que es más estable), 3 Sens *, a través del cruce entre sistemas . El 3 Sens * es lo que reacciona con el sustrato o con el 3 O 2 en los tres tipos de reacciones de fotooxigenación. [6]
![{\ displaystyle {\ ce {Sens -> [hv] {^ {1} Sens ^ {\ ast}} -> {^ {3} Sens ^ {\ ast}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6215cad829b7f70ab6da195d2ccda72164afd506)
Estados del oxígeno molecular

En las estructuras clásicas de Lewis , el oxígeno molecular, O 2 , se representa con un doble enlace entre los dos átomos de oxígeno. Sin embargo, los orbitales moleculares del O 2 son en realidad más complejos de lo que parecen sugerir las estructuras de Lewis. El orbital más alto ocupado (HOMO) de O 2 es un par de degenerado antienlazante π orbitales, π 2px * y π 2py *, que están tanto por separado ocupada por espín electrones desapareados. [4] Estos electrones son la causa de que el O 2 sea un diradical triplete en el estado fundamental (indicado como 3 O 2 ).
Si bien los HOMO de muchas moléculas estables consisten en unir orbitales moleculares y, por lo tanto, requieren un salto de energía moderado desde el enlace al antienlace para alcanzar su primer estado excitado, la naturaleza antienlazante del HOMO del oxígeno molecular permite una brecha de energía más baja entre su estado fundamental y el primer estado excitado. . Esto hace que la excitación de O 2 sea un proceso menos restrictivo energéticamente. En el primer estado excitado de O 2 , un aumento de energía de 22 kcal / mol desde el estado fundamental, ambos electrones en los orbitales antienlazantes ocupan un orbital π * degenerado, y el oxígeno ahora está en un estado singlete (indicado como 1 O 2 ). [3] 1 O 2es muy reactivo con una vida útil de entre 10 y 100 µs. [4]
Tipos de fotooxigenación
Los tres tipos de reacciones de fotooxigenación se distinguen por los mecanismos por los que proceden, ya que son capaces de producir productos diferentes o similares según las condiciones ambientales. Las reacciones de tipo I y II proceden a través de intermedios neutros, mientras que las reacciones de tipo III proceden a través de especies cargadas. La ausencia o presencia de 1 O 2 es lo que distingue las reacciones de tipo I y de tipo II, respectivamente. [1]
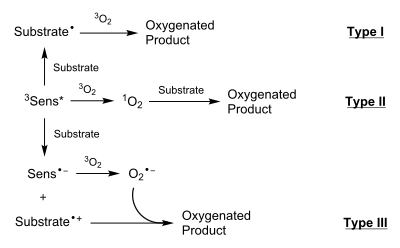
Tipo I
En las reacciones de tipo I, el 3 Sens * fotoactivado interactúa con el sustrato para producir un sustrato de radicales , generalmente a través de la ruptura del enlace homolítico de un enlace de hidrógeno en el sustrato. Este radical sustrato luego interactúa con 3 O 2 (estado fundamental) para producir un radical sustrato-O 2 . Generalmente, dicho radical se apaga extrayendo un hidrógeno de otra molécula de sustrato o del disolvente. Este proceso permite la propagación en cadena de la reacción.
Ejemplo: Atrapamiento de oxígeno de intermedios dirradicales
Las reacciones de fotooxigenación de tipo I se utilizan con frecuencia en el proceso de formación y captura de especies dirradicales . Mirbach y col. informó sobre una reacción de este tipo en la que un compuesto azo es lisado mediante fotólisis para formar el hidrocarburo dirradical y luego atrapado de forma escalonada por el oxígeno molecular: [7]

Tipo II
En las reacciones de tipo II, 3 Sens * transfiere su energía directamente con 3 O 2 a través de una transición sin radiación para crear 1 O 2 . Luego, el 1 O 2 se agrega al sustrato de diversas formas, entre las que se incluyen: cicloadiciones (más comúnmente [4 + 2]), adición de enlaces dobles para producir 1,2-dioxetanos y reacciones de eno con olefinas . [2]
Ejemplo: precursor de la síntesis de prostaglandinas
La cicloadición [4 + 2] de oxígeno singlete a ciclopentadieno para crear cis -2-ciclopenteno-1,4-diol es un paso común involucrado en la síntesis de prostaglandinas . [8] La adición inicial de oxígeno singlete, a través de la cicloadición concertada [4 + 2], forma un endoperóxido inestable . La posterior reducción del peróxido unido produce los dos grupos alcohol.

Tipo III
En las reacciones de tipo III, se produce una transferencia de electrones entre el 3 Sens * y el sustrato, lo que da como resultado un Sens aniónico y un sustrato catiónico . Entonces ocurre otra transferencia de electrones donde el aniónico Sens transfiere un electrón a 3 O 2 para formar el anión superóxido, O 2 - . Esta transferencia devuelve el Sens a su estado fundamental. El anión superóxido y el sustrato catiónico interactúan luego para formar el producto oxigenado.
Ejemplo: fotooxigenación con indolizina
La fotooxigenación de indolizinas (derivados aromáticos heterocíclicos del indol) se ha investigado tanto en contextos mecanicistas como sintéticos. En lugar de proceder a través de un mecanismo de fotooxigenación de Tipo I o Tipo II, algunos investigadores han optado por utilizar 9,10-dicianoantraceno (DCA) como fotosensibilizador, lo que lleva a la reacción de un derivado de indolizina con el radical anión superóxido. Tenga en cuenta que la reacción procede a través de un catión intermedio de radical indolizina que no se ha aislado (y por lo tanto no se muestra): [9]

Aplicaciones
Síntesis orgánica
Los 3 tipos de fotooxigenación se han aplicado en el contexto de la síntesis orgánica. En particular, las fotooxigenaciones de tipo II han demostrado ser las más utilizadas (debido a la baja cantidad de energía necesaria para generar oxígeno singlete) y se han descrito como "uno de los métodos más potentes para la oxifuncionalización fotoquímica de compuestos orgánicos". [10] Estas reacciones pueden ocurrir en todos los solventes comunes y con una amplia gama de sensibilizadores.
Muchas de las aplicaciones de las fotooxigenaciones de tipo II en la síntesis orgánica provienen de las investigaciones de Waldemar Adam sobre la reacción enérgica del oxígeno singlete con alquenos acíclicos. [10] A través del efecto cis y la presencia de grupos de dirección apropiados, la reacción puede incluso proporcionar una alta regioselectividad y diastereoselectividad , dos valiosos controles estereoquímicos. [11]

Terapia fotodinámica
La terapia fotodinámica (TFD) utiliza fotooxigenación para destruir el tejido canceroso . [12] Se inyecta un fotosensibilizador en el tumor y luego se exponen longitudes de onda de luz específicas al tejido para excitar la Sens. La Sens excitada generalmente sigue un mecanismo de fotooxigenación de tipo I o II para producir daño oxidativo en las células. El daño oxidativo extenso a las células tumorales destruirá las células tumorales. Además, el daño oxidativo a los vasos sanguíneos cercanos provocará una aglomeración local y cortará el suministro de nutrientes al tumor, lo que hará que el tumor se muera de hambre. [13]
Una consideración importante al seleccionar el Sens para usar en PDT es la longitud de onda de luz específica que el Sens absorberá para alcanzar un estado excitado. Dado que la penetración máxima de los tejidos se logra alrededor de longitudes de onda de 800 nm, seleccionar Sens que absorban alrededor de este rango es ventajoso ya que permite que la TFD sea efectiva en los tumores debajo de la capa más externa de la dermis. La ventana de luz de 800 nm es más eficaz para penetrar los tejidos porque a longitudes de onda inferiores a 800 nm la luz comienza a ser dispersada por las macromoléculas de las células y a longitudes de onda superiores a 800 nm las moléculas de agua comienzan a absorber la luz y convertirla en calor. . [4]
Referencias
- ↑ a b IUPAC (1997). AD McNaught y A. Wilkinson (ed.). Compendio de terminología química . Publicaciones científicas de Blackwell, Oxford. doi : 10.1351 / goldbook . ISBN 978-0-9678550-9-7.
- ^ a b c d M. R. Iesce; et al. (2005). "Fotooxigenación de heterociclos". Curr. Org. Chem . 9 (2): 109-139. doi : 10.2174 / 1385272053369222 .
- ↑ a b c C.S. Foote (1968). "Mecanismos de oxidación fotosensibilizada". Ciencia . 162 (3857): 963–970. Código Bibliográfico : 1968Sci ... 162..963F . doi : 10.1126 / science.162.3857.963 .
- ↑ a b c d e I. J. MacDonald y TJ Dougherty (2001). "Principios básicos de la terapia fotodinámica". Revista de porfirinas y ftalocianinas . 5 (2): 105-129. doi : 10.1002 / jpp.328 .
- ^ La mayor parte de la literatura reciente (posterior a 2000) incluye la clasificación "Tipo III"; sin embargo, los artículos más antiguos solo reconocen el Tipo I y el Tipo II como clases denominadas de reacciones de fotooxigenación.
- ^ CS Foote (1987). Mecanismos de acción fotodinámica tipo I y tipo II . Serie de simposios ACS . 339 . págs. 22–38. doi : 10.1021 / bk-1987-0339.ch002 . ISBN 978-0-8412-1026-4.
- ^ Mirbach, Marlis; M. Manfred; A. Saus (1982). "Fotoquímica de alta presión y espectroscopia ultravioleta en sistemas gas-líquido". Revisiones químicas . 82 (1): 59–76. doi : 10.1021 / cr00047a003 .
- ^ Cigüeña, Gilbert; P. Sher; H. Chen (octubre de 1986). "Ciclización radical-atrapamiento en la síntesis de productos naturales. Una ruta simple, estereocontrolada para prostaglandina Fza". Mermelada. Chem. Soc . 108 (20): 6384–6385. doi : 10.1021 / ja00280a043 .
- ^ Li, Yun; H. Hu; J. Ye; H. Diversión; H. Hu; J. Xu (2004). "Modos de reacción y mecanismo en reacciones de fotooxigenación de indolizina". Revista de Química Orgánica . 69 (7): 2332–2339. doi : 10.1021 / jo035070d .
- ↑ a b Rumbach, editado por Jochen Mattay y Axel G. Griesbeck en cooperación con Christian Stammel, Joachim Hirt y Thomas (1994). Jochen Mattay y Axel Griesbeck (ed.). Pasos fotoquímicos clave en la síntesis orgánica: un libro de curso experimental . Weinheim: VCH. ISBN 978-3-527-29214-1.CS1 maint: texto adicional: lista de autores ( enlace )
- ^ Adán, Waldemar; W. Bruenker (1993). "Fotooxigenación diastereoselectiva y regioselectiva de una amina alílica quiral y sus derivados de acilo: evidencia estereoquímica de un efecto de dirección por parte del grupo amino en la reacción eno del oxígeno singlete". Mermelada. Chem. Soc . 115 (7): 3008–3009. doi : 10.1021 / ja00060a072 .
- ^ Dougherty, Thomas (mayo de 1987). "Fotosensibilizadores: terapia y detección de tumores malignos". Fotoquímica y Fotobiología . 45 (445): 879–889. doi : 10.1111 / j.1751-1097.1987.tb07898.x .
- ^ Chen, Qun; Z. Huang; H. Chen; H. Shapiro; J. Beckers; F. Hetzel (agosto de 2002). "Mejora de la respuesta tumoral mediante la manipulación de la oxigenación del tumor durante la terapia fotodinámica". Fotoquímica y Fotobiología . 76 (2): 197–203. doi : 10.1562 / 0031-8655 (2002) 0760197IOTRBM2.0.CO2 .